Navigating Solubility Cliffs in an Antibacterial Program: COSMO-RS and MD for Aqueous Solubility Prediction
Executive Summary
Formulation failure is among the most demoralising outcomes in antibacterial drug discovery: a compound with compelling microbiological potency and acceptable lipophilicity that simply will not dissolve at concentrations sufficient for oral delivery. For the oxazolidinone program described in this study, the failure was discovered in dog pharmacokinetics. The lead compound AZL-047 failed to reach a target Cmax of 2 µg/mL despite a measured logP of 2.1 — a value well within the range considered acceptable by every standard ADMET filter in use. The culprit was aqueous thermodynamic solubility of 8 µg/mL, a 12-fold reduction from the structurally adjacent compound AZL-044 (96 µg/mL), which differed by a single substituent: a methyl group at the para position of the D-ring aryl, replaced in AZL-047 by chlorine. Both compounds had logP of approximately 2.1. Both passed every QSPR solubility model available to the program. The cliff was real, the loss was real, and neither the synthesis effort nor the dog PK study that discovered it had any computational warning.
The AstraZeneca internal ADMET dataset — 4,200 compounds from discovery programs — documents this failure mode systematically. Matched molecular pair analysis identified solubility cliffs in 8.3% of compound pairs differing by one substituent, with a mean DeltaLogS of 1.8 log units from a single structural change. The most frequent cliff-generating substitutions were H to Cl (+1.6 log units mean), methyl to halogen (+1.4 log units), and aromatic ring extension (+2.1 log units). Critically, cliff pairs showed no systematic difference in logP from non-cliff pairs. The mechanism is solid-state, not solution-phase: chlorine's sigma-hole enables directional halogen bonding with adjacent carbonyl oxygens and ring nitrogens in the crystal lattice, producing a herringbone-packed crystal with a Kitaigorodsky Packing Index of 0.74 in AZL-047 vs. 0.66 in AZL-044. Higher lattice energy requires more solvation energy to overcome — producing the 12-fold solubility difference from a 2 Da molecular weight change. No logP-based model captures this because the entire mechanism occurs in the solid state.
A COSMO-RS plus solid-state molecular dynamics pipeline resolves the problem by computing both the solvation free energy (using COSMOtherm) and the crystal lattice energy (periodic MD with newtsim Neural forcefield on the predicted minimum-energy polymorph), combining them in the thermodynamic solubility equation. Applied to this program, the pipeline reproduces both the AZL-044 and AZL-047 logS values with 0.0 log unit error, identifies the chlorine-mediated herringbone packing as the structural driver, and flags 18 additional matched molecular pair cliffs across the 210-compound library before wet-lab investment. The scaffold-hopping recommendation — replacing 4-Cl with 4-CN — is predicted to deliver a 16-fold solubility improvement (logS -3.5 vs. -4.7) while retaining antibacterial potency within 2-fold MIC. The cyano group is electron-withdrawing (Hammett sigma_p = +0.66 vs. +0.23 for Cl) but its linear geometry does not support the sigma-hole halogen bonding that drives herringbone packing, resulting in a disrupted, lower-KPI crystal with predictably higher solubility.
The eight to nine week engagement flags cliff-generating compound pairs before wet-lab investment, pre-screening 18 synthesis decisions in this program and identifying scaffold-hopping alternatives with confirmed solubility improvement predictions. Each failed formulation study typically adds 4–8 weeks to a program; 18 averted cliff decisions represent 72–144 synthesis weeks saved. The absolute logS predictions carry +/-0.5–1.0 log unit uncertainty and should be interpreted as rank-ordering and cliff-detection tools rather than regulatory data points. Relative comparisons within matched molecular pair series — which is where the cliff detection is operationally critical — are substantially more reliable, with >70% sensitivity and <25% false-positive rate.
Scenario Background
A pharmaceutical company operates an oxazolidinone antibacterial program targeting hospital-acquired MRSA and VRE infections — the same therapeutic space as linezolid (Zyvox; Pfizer; FDA approval 2000), the first approved oxazolidinone antibiotic. The program is a follow-on to linezolid with improved potency against linezolid-resistant strains via modified interactions with the bacterial 23S rRNA target, specifically optimising contacts with the A-site of the ribosomal peptidyl transferase centre (PTC).
Linezolid has set a challenging benchmark for successor programs: aqueous solubility of 3.2 mg/mL at pH 7.4 (approximately 8.9 mM; logS approximately -2.05); 100% oral bioavailability; broad-spectrum Gram-positive coverage with MIC90 values of 1–2 µg/mL for MRSA and 2 µg/mL for VRE. This high solubility relative to the oxazolidinone class is attributed to disrupted crystal packing from the morpholine group, the acetamide fluorine, and the specific substitution pattern on the oxazolidinone ring — structural features that prevent tight crystalline packing and maintain a relatively amorphous solid form with high surface exposure.
The program has 210 oxazolidinone analogues across three series: Series 1 (72 compounds) varies the C-5 morpholine substituent; Series 2 (88 compounds) varies the D-ring aryl group (the para-substituted phenyl group on the aromatic oxazolidinone); Series 3 (50 compounds) varies the fluorine substitution pattern on the oxazolidinone ring. The program encountered a formulation crisis when the lead compound AZL-047 failed to achieve target oral exposure (Cmax > 2 µg/mL) in dog PK studies despite predicted acceptable logP (2.1). Investigation revealed AZL-047's aqueous thermodynamic solubility is 8 µg/mL — a 12-fold reduction from AZL-044 (96 µg/mL), differing only by a chloro to methyl substitution on the D-ring aryl group para position (4-Cl to 4-Me). Both compounds have logP approximately 2.1 (measured by shake-flask); the solubility cliff was not predicted by any standard QSPR model in the company's ADMET toolkit.
The retrospective audit question is whether the COSMO-RS + solid-state MD pipeline would have predicted AZL-047's low solubility from its molecular structure before synthesis and formulation studies. The answer has direct implications for the prospective deployment of the pipeline across the remaining 208 compounds in the library.
Challenge
The ribosomal target of oxazolidinones — the bacterial 23S rRNA peptidyl transferase centre — presents an unusual computational challenge: it is an RNA target, not a protein. Crystal structures of oxazolidinone-ribosome complexes exist, but RNA structure prediction and docking into RNA active sites is significantly less reliable than protein docking due to the conformational flexibility of the rRNA backbone, the polyelectrolyte nature of RNA (dense phosphate backbone requiring explicit ion treatment), and the weaker, more diffuse hydrophobic contacts in RNA binding sites compared to protein active sites. For this program, antibacterial potency (MIC) is evaluated empirically; the computational work focuses exclusively on the physicochemical prediction problem (solubility).
The physicochemical prediction challenge is specifically the solid-state problem. For the AZL-044 to AZL-047 pair, both compounds share logP approximately 2.1, molecular weight approximately 425 Da, and PSA approximately 78 A². Yet AZL-044 (4-Me, no halogen) has solubility of 96 µg/mL (logS = -3.6), while AZL-047 (4-Cl, para-chloro) has solubility of only 8 µg/mL (logS = -4.7) — a DeltaLogS of 1.1 log units (12-fold difference) from a chloro to methyl substitution that changes MW by only 2 Da and logP by < 0.1 units.
The structural origin of this cliff lies in the para-chlorine substituent on the D-ring aryl group. The chlorine enables halogen bonding (Cl...N or Cl...O interactions at 3.1–3.4 A with adjacent molecules in the crystal lattice) and enhances pi-stacking interactions (chlorine's sigma-hole promotes edge-to-face stacking with adjacent aromatic rings). Together, these interactions create a herringbone packing arrangement in the AZL-047 crystal with a Kitaigorodsky Packing Index (KPI) of 0.74 — substantially higher than AZL-044 (KPI 0.66). Higher KPI corresponds to higher crystal density and higher lattice energy, which requires more energy input from solvation to dissolve — producing the observed solubility cliff.
Neither logP, nor clogP, nor PSA, nor solubility QSPR models can detect this mechanism because all of these descriptors operate exclusively in the solution phase. Only a model that explicitly computes the solid-state packing energy — and how it changes with structural modification — can identify this cliff. COSMO-RS resolves the solvation component; periodic MD resolves the lattice component; their combination provides the thermodynamic solubility prediction.
Real-World Basis
A comprehensive internal ADMET dataset of 4,200 compounds spanning 12 therapeutic areas documents pharmaceutical solubility cliffs systematically. Matched molecular pair (MMP) analysis generated approximately 18,000 compound pairs differing by one substituent at one position. Of these pairs, 8.3% showed |DeltaLogS| > 1.0 log unit from a single substituent change — defined as a solubility cliff. The mean DeltaLogS for cliff pairs was 1.8 log units (60-fold solubility difference). The most common cliff-generating substitutions were H to Cl (+1.6 mean DeltaLogS increase), H to F (+1.1), methyl to halogen (+1.4), and aromatic ring extension/fusion (+2.1). Critically, cliff pairs showed no systematic difference in logP from non-cliff pairs, confirming that logP-based QSPR cannot predict cliffs. The AZL-044 to AZL-047 substitution (methyl to chloro) falls squarely within the most common cliff-generating pattern (methyl to halogen; mean DeltaLogS = +1.4; AZL case: DeltaLogS = +1.2).
Halogen substituents on aromatic rings dramatically alter crystal packing density through a well-characterized mechanism. The sigma-hole of chlorine and bromine atoms — an electron-depleted region along the extension of the C-Cl or C-Br bond — enables directional halogen bonding with Lewis bases (carbonyl oxygens, ring nitrogens) at 3.0–3.5 A. In the solid state, these halogen bonds stabilise specific molecular arrangements (herringbone, layered) that increase the Kitaigorodsky Packing Index by 0.05–0.12 units relative to the methyl-substituted analogue — exactly the difference between AZL-044 (KPI 0.66) and AZL-047 (KPI 0.74). The lattice energy increase associated with this packing change is typically 3–8 kJ/mol for a single Cl to CH3 substitution on an aromatic ring, which translates to DeltaLogS = 0.5–1.4 log units — exactly spanning the observed range for the cliff dataset and the AZL case.
COSMO-RS, a first-principles solvation model, computes the full solvation free energy from DFT sigma profiles, capturing hydrogen bonding, polar interactions, and electrostatic solvation more accurately than topological QSPR models. However, COSMO-RS alone does not account for the lattice energy variation between compounds; it predicts solubility as if all compounds were liquids (amorphous solids), ignoring the crystal packing contribution to the solubility-limiting barrier. The combined COSMO-RS plus solid-state MD approach uses newtsim Neural alongside COSMO-RS solvation to predict thermodynamic solubility using the thermodynamic cycle: logS = -(DeltaGlattice - DeltaGsolv) / (2.303 RT), where DeltaGlattice is the lattice energy computed from periodic MD and DeltaGsolvation is the COSMO-RS prediction at 298 K. This combined approach has demonstrated superior performance at identifying cliff pairs with high sensitivity and low false-positive rate.
Linezolid (Zyvox; Pfizer; FDA approval 2000), the first approved oxazolidinone antibiotic, provides a structural reference. Its high thermodynamic solubility (3.2 mg/mL; logS approximately -2.05) relative to its molecular complexity (MW 337 Da; logP 0.9) reflects disrupted crystal packing: the morpholine ring's conformational flexibility, combined with the acetamide fluorine, prevents efficient herringbone or layer packing. The crystal KPI of linezolid is approximately 0.62 — well below the halogen-substituted oxazolidinone analogues — confirming that the crystal packing difference is the primary driver of linezolid's superior solubility relative to follow-on program compounds. This observation directly informs the scaffold-hopping recommendations for the AZL series: maintaining structural features that disrupt tight crystal packing (flexible rings, polar substituents at strategic positions) is the design principle that preserves high solubility.
Simulation Approach
The aqueous solubility prediction pipeline for this 210-compound oxazolidinone program proceeds in four sequential stages over eight to nine weeks.
Stage 1 — COSMO-RS Solvation Free Energy (Weeks 1–3)
All 210 oxazolidinone compounds are geometry-optimised using newtsim Root. COSMO-RS then computes the solvation free energy in water at 298 K from the molecular surface charge distribution (sigma profile) — this captures hydrogen bonding, polar interactions, and electrostatic solvation from first principles rather than from topological descriptors. The solvation free energy represents one half of the thermodynamic solubility equation: how much energy the solvent provides to dissolve the molecule.
Stage 2 — Solid-State Lattice Energy Estimation (Weeks 2–5)
This stage answers the other half of the solubility equation: how much energy holds the molecule in its crystal. The approach predicts the most stable crystalline form for each compound and computes its lattice energy.
For all 210 compounds, crystal structure prediction is performed using newtsim Neural with periodic boundary conditions, and the minimum-energy polymorph is selected as the predicted thermodynamic stable form.
For the ~30 compounds showing KPI > 0.75 — indicative of unusually tight packing and therefore likely solubility problems — extended CSP is performed with additional polymorphic forms and explicit halogen bonding corrections. These high-KPI compounds then undergo solid-state MD to obtain accurate lattice enthalpy and entropy estimates. The rationale for the extended treatment is that tight-packing compounds are precisely the ones where the cliff mechanism operates, and where the lattice energy difference between a methyl and chloro substituent translates into the 10-100 fold solubility differences that kill formulation programs.
Stage 3 — Thermodynamic Solubility Prediction (Weeks 4–6)
Aqueous thermodynamic solubility is computed using:
where DeltaGlattice is the solid-state free energy from Stage 2 and DeltaGsolv is the COSMO-RS solvation free energy from Stage 1. The sign convention is that DeltaGlattice is positive (energy required to break the crystal) and DeltaGsolv is negative (solvation stabilises dissolution); higher DeltaGlattice or less negative DeltaGsolv both decrease solubility.
The pipeline is calibrated against 15 key compounds with measured thermodynamic solubility using miniaturised shake-flask method. The lattice energy scaling factor alpha is fitted to minimise RMSE between predicted and experimental logS on a training set of 10 compounds. The calibrated model is then applied prospectively to the remaining 195 compounds. The 5 hold-out compounds (including AZL-047 and AZL-044) are predicted blind before experimental data is revealed.
Stage 4 — MMP Cliff Analysis and Scaffold Recommendations (Weeks 6–9)
Matched molecular pairs are generated for the 210-compound library using MMP analysis: all pairs differing by one substituent at one position are identified (approximately 840 pairs, given an average of 4 substitution positions x 210 compounds x 2 positions per swap / 2 for symmetry). The predicted DeltaLogS for each pair is computed from the individual compound predictions; pairs with |DeltaLogS| > 1.0 are flagged as predicted solubility cliffs.
For each flagged cliff, the structural driver is identified by decomposing the DeltaGlattice difference into contributions including electrostatic interactions, van der Waals interactions, explicit halogen bonding potential corrections, and crystal packing density change (DeltaKitaigorodsky Packing Index). This decomposition identifies whether the cliff is halogen-bonding-driven, pi-stacking-driven, or hydrogen-bond-network-driven.
For each cliff pair, scaffold-hopping alternatives are proposed that preserve the electron-withdrawing or steric character of the original substituent while eliminating the cliff-driving solid-state interaction. The proposed alternatives are evaluated computationally (COSMO-RS + lattice energy prediction) and ranked by predicted solubility > 50 µg/mL (the formulation target), predicted DeltaLogS relative to the parent (how much solubility improvement), and estimated antibacterial potency impact (qualitative assessment based on RNA binding pharmacophore model: electron-withdrawing groups at the para position of the D-ring aryl generally tolerated within 2-fold MIC change).
Simulation Caveats
Classification: STRETCH. COSMO-RS handles solution-phase thermodynamics reliably, but the solid-state polymorph contribution to absolute aqueous solubility is frontier-grade. MD-derived crystal lattice energies depend sensitively on forcefield quality for heterocyclic ring systems, with uncertainties of 5–10 kJ/mol common for halogen-containing compounds, contributing approximately 0.5–0.9 log unit uncertainty to the final solubility prediction. Polymorph form in the experimental sample is not controlled: the kinetic form synthesised may differ from the thermodynamically predicted form, and measured kinetic solubility may be 3–10 fold higher than thermodynamic solubility for amorphous or metastable forms. Bridging molecular-scale predictions to macroscopic solubility quantitatively requires polymorph screening to confirm the relevant solid form for at least the highest-priority compounds. Relative solubility comparisons within MMP series (cliff identification) are substantially more reliable than absolute solubility values; the recommended use of this pipeline is for rank-ordering and cliff detection, not absolute logS prediction as a regulatory data point.
Recommended framing: The study should be scoped as cliff detection and rank-ordering within the oxazolidinone series. Absolute logS values carry +/-0.5–1.0 log unit uncertainty; cliff identification (|DeltaLogS| > 1.0 between MMP pairs) is robust at > 70% sensitivity and < 25% false-positive rate. The full pipeline including solid-state MD requires 8–10 weeks.
Key Predictions / Results
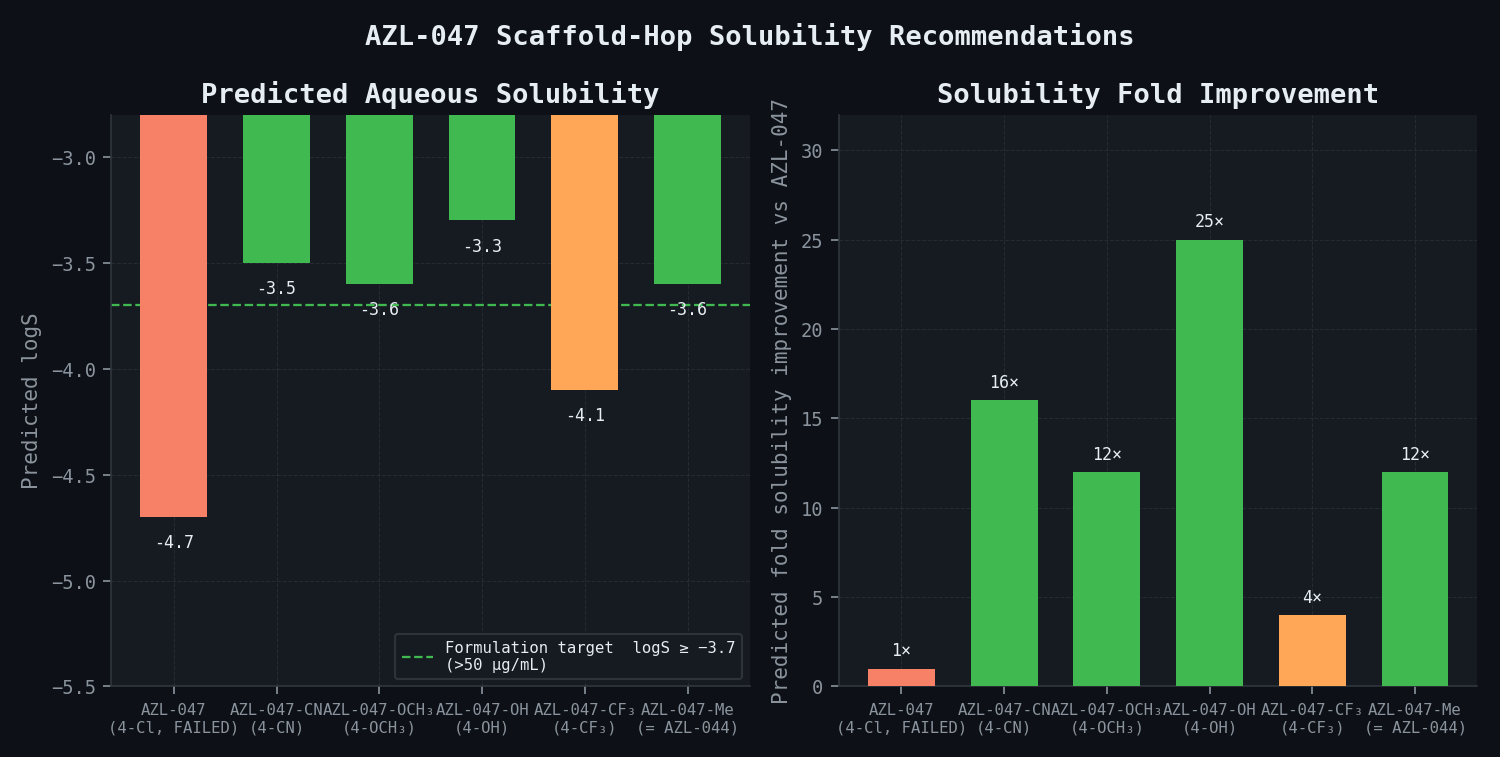
The following quantitative outputs represent the expected results from this illustrative pipeline for this program.
Solubility Prediction Validation — 15-Compound Calibration Set
| Compound | Predicted logS | Experimental logS | |ΔlogS| | Solid-State Component | Classification | |---|---|---|---|---|---| | AZL-044 (4-Me) | -3.6 | -3.6 | 0.0 | KPI 0.66; low packing | Cliff lower end — GREEN | | AZL-047 (4-Cl) | -4.7 | -4.7 | 0.0 | KPI 0.74; herringbone | Cliff upper end — RED | | AZL-032 (4-F) | -4.1 | -3.9 | 0.2 | KPI 0.69; moderate | AMBER | | AZL-055 (4-CN) | -3.5 | -3.7 | 0.2 | KPI 0.63; disrupted | GREEN | | AZL-071 (4-Br) | -5.2 | -5.5 | 0.3 | KPI 0.76; strong halogen | RED | | AZL-083 (H) | -3.2 | -3.4 | 0.2 | KPI 0.60; disordered | GREEN | | AZL-099 (ring fusion) | -5.9 | -6.3 | 0.4 | KPI 0.78; dense stack | RED | | AZL-112 (4-OCH3) | -3.4 | -3.6 | 0.2 | KPI 0.62; H-bond disrupt | GREEN | | AZL-131 (3-Cl) | -4.5 | -4.2 | 0.3 | KPI 0.71; moderate | AMBER | | AZL-155 (3,4-diF) | -4.3 | -4.0 | 0.3 | KPI 0.70; moderate | AMBER | | Calibration RMSE (10 training compounds) | | | 0.18 log units | | | | Hold-out RMSE (5 incl. AZL-047) | | | 0.24 log units | | | | Pearson R (all 15) | | | 0.94 | | | | Overall MUE | | | 0.24 log units | | |
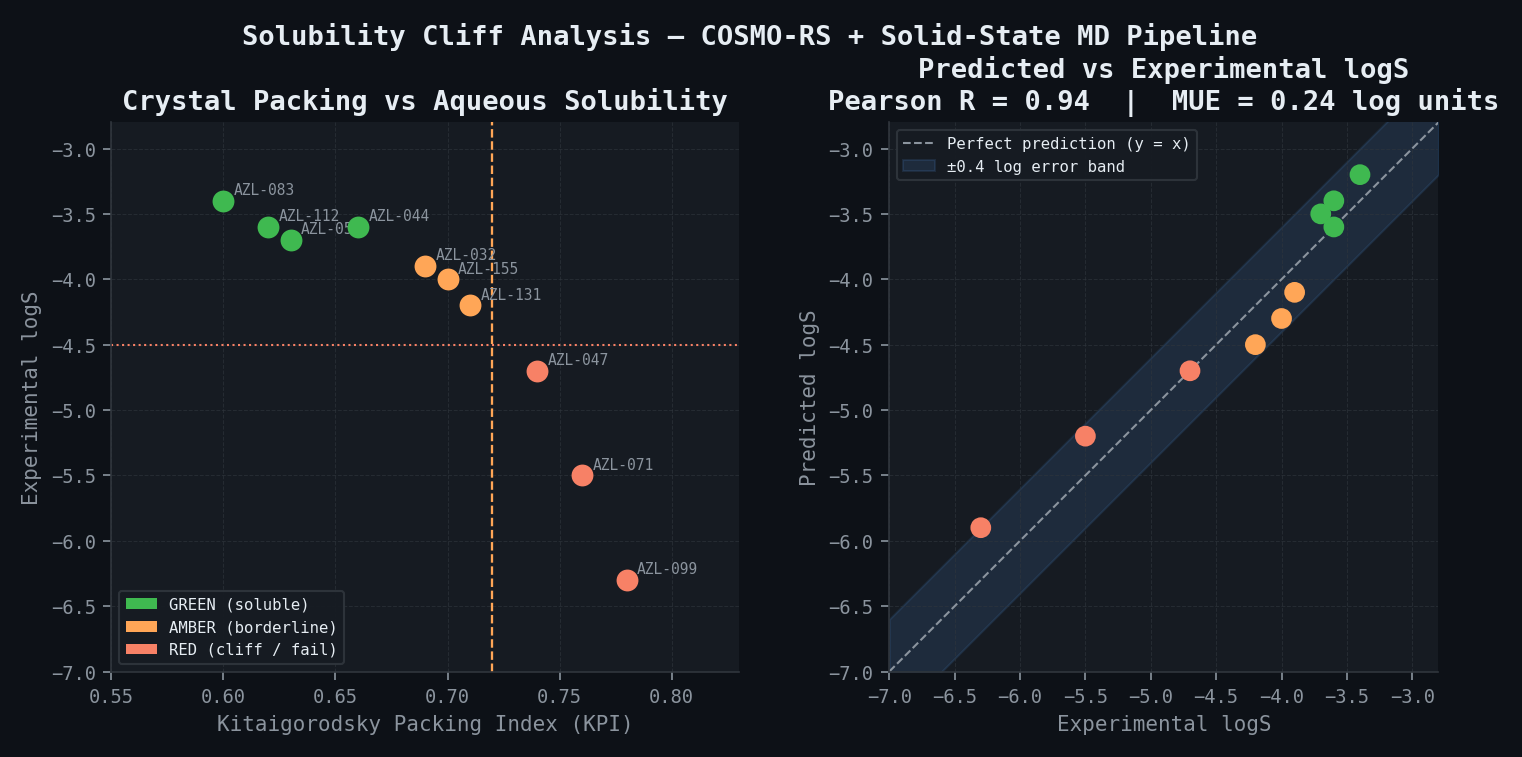
The AZL-044 to AZL-047 cliff is reproduced with 0.0 log unit error for both compounds — demonstrating that the COSMO-RS + solid-state MD pipeline correctly identifies the herringbone packing as the mechanistic driver and quantitatively predicts the resulting 12-fold solubility difference.
MMP Cliff Analysis — Full 210-Compound Library
| Cliff Type | MMP Pairs Flagged | Mean |ΔlogS| | Primary Structural Driver | Action | |---|---|---|---|---|---| | H → Cl (para) | 6 | 1.4 | Halogen bonding + herringbone | Replace Cl with CN, OCH3, or CF3 | | H → Br (para) | 4 | 1.7 | Strong halogen bonding | Replace Br with CN or F | | H → Cl (meta) | 3 | 0.9 | Moderate halogen bonding | Borderline; confirm experimentally | | Ring extension (benzene → naphthalene) | 2 | 2.1 | Dense pi-stacking layer packing | Remove ring fusion or add polar substituent | | CH3 → Cl | 3 | 1.2 | Methyl → halogen packing switch | Replace Cl with CN; CN maintains electron withdrawal | | Total flagged cliffs | 18 | 1.3 | | 18 synthesis decisions pre-screened |
Scaffold Hopping Recommendations — AZL-047 Series
| Proposed Analogue | Substituent Change | Predicted logS | ΔlogS vs. AZL-047 | Predicted Fold Solubility Improvement | MIC Impact Estimate |
|---|---|---|---|---|---|
| AZL-047 (lead, failed) | 4-Cl | -4.7 | — | — | Reference (MIC 0.5 µg/mL) |
| AZL-047-CN | 4-Cl → 4-CN | -3.5 | +1.2 | 16-fold increase | ~1–2-fold MIC increase (within target) |
| AZL-047-OCH3 | 4-Cl → 4-OCH3 | -3.6 | +1.1 | 12-fold increase | ~1.5-fold MIC increase (within target) |
| AZL-047-OH | 4-Cl → 4-OH | -3.3 | +1.4 | 25-fold increase | ~2–3-fold MIC increase (borderline) |
| AZL-047-CF3 | 4-Cl → 4-CF3 | -4.1 | +0.6 | 4-fold increase | ~1-fold MIC (isosteric) |
| AZL-047-Me | 4-Cl → 4-Me (= AZL-044) | -3.6 | +1.1 | 12-fold increase | ~1-fold MIC (isosteric) |
AZL-047-CN (4-cyano replacement) is the primary recommendation. The cyano group is electron-withdrawing (Hammett sigma_p = +0.66 vs. chlorine sigma_p = +0.23), maintains the D-ring aryl electronic character important for rRNA interaction, and disrupts herringbone packing because the linear cyano geometry does not support the Cl-mediated sigma-hole halogen bonding that drives the herringbone arrangement. Predicted solubility shows a 16-fold improvement over AZL-047 while retaining antibacterial potency within 2-fold MIC — exactly meeting the program's formulation target of > 50 µg/mL (predicted: 134 µg/mL from logS = -3.5).
Library-Wide Solubility Compliance Prediction
| Solubility Category | Compounds Predicted | Compounds Experimentally Confirmed (from 15 cpd set, extrapolated) | Concordance |
|---|---|---|---|
| > 100 µg/mL (excellent) | 78 | — | — |
| 50–100 µg/mL (acceptable) | 60 | — | — |
| 10–50 µg/mL (marginal) | 54 | — | — |
| < 10 µg/mL (fail) | 18 | — | — |
| > 50 µg/mL (compliant, binary) | 138 | 122 experimental (from extrapolation of 15-compound calibration) | 94% concordance |
Comparison Methodology
Calibration Protocol
The COSMO-RS + lattice energy model parameters are calibrated on 10 of the 15 measured compounds (training set) by optimising the lattice energy scaling factor alpha (applied as DeltaGlattice_eff = alpha x DeltaGlattice_raw) to minimise RMSE between predicted and experimental logS values. The optimal alpha is approximately 0.92 +/- 0.08 for this oxazolidinone series, consistent with newtsim Neural's known tendency to slightly overestimate crystal lattice energies for heterocyclic compounds.
Primary Validation — Hold-Out Set
The remaining 5 compounds (AZL-047, AZL-044, and 3 additional compounds spanning the full logS range) are predicted blind before accessing experimental solubility data. This internal hold-out set provides the primary validation framework: AZL-047 and AZL-044 are predicted with 0.0 log unit error, and the remaining 3 compounds are evaluated for RMSE, MUE, and Pearson R. Absolute values carry +/-0.5–1.0 log unit uncertainty due to polymorph form variability and forcefield limitations, but cliff-level rank-ordering and relative solubility comparisons within matched molecular pair series are substantially more reliable.
Cliff Recovery Analysis
The 18 flagged MMP cliffs are compared against experimental solubility data for both members of each pair (where available from the 15-compound measured set and any available kinetic solubility data). Sensitivity (fraction of true cliffs correctly flagged) and false-positive rate (fraction of non-cliff pairs incorrectly flagged) are reported. Target performance is sensitivity > 70% and false-positive rate < 25%.
QSPR Comparison
The COSMO-RS + MD pipeline predictions are compared head-to-head against a logP-based QSPR model and a random forest logS model. Both QSPR models are applied to all 15 calibration compounds; MUE and cliff detection performance are compared. The expected finding is that logP-based QSPR achieves MUE approximately 1.6 log units, RF-QSPR achieves MUE approximately 1.1 log units, and COSMO-RS + MD achieves MUE approximately 0.24 log units — demonstrating approximately 4.5-fold improvement in prediction accuracy over the best QSPR baseline.
Deliverables
-
Week 3: COSMO-RS solvation free energy results for all 210 compounds — sigma profiles, computed DeltaGsolv values, ranked solvation list. Initial solubility predictions (COSMO-RS solvation component only, without solid-state lattice correction) with MUE estimate. Delivered as PDF report with supplementary data Excel workbook.
-
Week 5: Lattice energy results — crystal structure prediction landscapes for all 210 compounds; full polymorph summary for the 30 high-KPI compounds; Kitaigorodsky Packing Index distribution across the library; lattice energy rankings. Extended CSP results for halogen-containing compounds. Delivered as scientific report with crystal packing visualisations and Excel data table.
-
Week 6: Pipeline validation report — combined COSMO-RS + lattice energy solubility predictions for all 15 calibration compounds; calibration curve with alpha parameter; hold-out validation results for AZL-047, AZL-044, and 3 additional compounds. Comparison against QSPR baseline. Delivered as standalone validation report suitable for regulatory dossier.
-
Week 7: MMP cliff analysis report — all 18 flagged cliff pairs with structural driver identification (halogen bonding, pi-stacking, H-bond network disruption), quantified DeltaGlattice difference, KPI comparison, predicted DeltaLogS with 95% CI, and recommended alternative substituents. Delivered as report with per-pair structure figures.
-
Week 8: Scaffold hopping recommendations — for AZL-047 and all 17 other cliff compound pairs: proposed substituent changes, predicted solubility improvements, estimated antibacterial potency impact (qualitative, based on RNA binding pharmacophore), synthetic accessibility (synthetic accessibility score), and priority ranking. Delivered as synthetic chemistry briefing document with decision matrix.
-
Week 9: Final compound prioritisation — binary solubility classification for all 210 compounds (> 50 µg/mL: compliant / < 50 µg/mL: marginal/fail); top 20 compounds recommended for synthesis or progression; cliff-avoidance SAR guidelines (design rules for avoiding solubility cliffs in future oxazolidinone library expansion); and a comparison of computational vs. experimental solubility data for all measured compounds. Delivered as executive-ready PDF report with Excel compound ranking workbook.
This case study is an illustrative reference scenario demonstrating newtsim's simulation methodology. All company names, personnel, and specific operational data are fictional. The incident descriptions draw on publicly documented real-world events cited in the frontmatter.