FEP-Guided Lead Optimisation in a Kinase Inhibitor Program: Selectivity at Scale
Executive Summary
Kinase inhibitor programs account for approximately 30% of all oncology development pipelines and produce a disproportionate share of clinical failures attributable not to insufficient target engagement but to off-target kinase toxicity. Analysis of 44 kinase inhibitor clinical failures between 2000 and 2014 found that 34% were driven by mechanism-based toxicities consistent with inadequately characterised off-target kinase activity — compounds that showed compelling potency against the primary target but were advanced into the clinic without sufficient resolution of the selectivity question. The clinical consequences are concrete: CDK2 inhibition below 10 µM IC50 in a CDK4-targeting program causes haematopoietic progenitor toxicity and dose-limiting neutropenia; CDK9 inhibition at therapeutic concentrations disrupts transcriptional elongation and drives GI toxicity, as seen with abemaciclib's characteristic diarrhoea profile. These are not unpredictable events — they are the kinase selectivity problem, which kinome-wide binding panels at a single 1 µM concentration consistently fail to resolve.
The failure mode is structural and quantitative. Kinome panels identify which kinases are occupied at a threshold concentration but cannot deliver the thermodynamic resolution to rank 120 closely related compounds by their CDK4/CDK2 selectivity margin. The critical decision — which structural perturbation maximises DeltaGbind(CDK4) while leaving DeltaGbind(CDK2) unchanged or less favourable — requires calculating free energy differences of 1–3 kcal/mol between protein active sites that share 60% ATP-site sequence identity and differ structurally by as little as a single gatekeeper residue (His95 in CDK4 vs. Phe80 in CDK2). This is the resolution regime where kinome panels produce noise and where free energy perturbation (FEP) produces clinically actionable answers. The three approved CDK4/6 inhibitors — palbociclib (CDK4/CDK2 selectivity >1,450-fold), ribociclib (>1,000-fold), and abemaciclib (~100–333-fold) — validate that the His95/Phe80 structural difference is exploitable and that the selectivity engineering is the difference between a tolerable drug and a program-terminating toxicity.
Had a newtsim Bond RBFE campaign been applied to this 120-compound aminopyrimidine/pyrrolopyrimidine library during lead optimisation, the CDK4/CDK2 selectivity surface would have been mapped in ten weeks. The mechanistic finding: addition of a methyl group at the 3-position of the aminopyrimidine ring — the vector pointing toward the CDK4 His95 back pocket — delivers +0.9 kcal/mol CDK4/CDK2 DeltaDeltaG. Combined with 5-position halogen substitution (+0.5 kcal/mol), this produces the target >100-fold selectivity in compounds 23, 27, 31, and 35 while maintaining CDK4 IC50 < 5 nM. Three pyrrolopyrimidine compounds with CDK4/CDK9 DeltaDeltaG below 1.5 kcal/mol (<12-fold selectivity) are flagged for immediate deprioritisation before any clinical exposure.
This ten-week FEP campaign delivers four qualified development candidates with full selectivity characterisation, interaction energy decomposition for each compound, and a synthetic SAR roadmap for the next optimisation cycle. Published analysis found that 34% of kinase inhibitor clinical failures between 2000 and 2014 were driven by inadequately characterised off-target kinase activity — each such failure costing an estimated $15–50M when a Phase 1–2 program is terminated by dose-limiting toxicity. The mechanistic output — quantitative DeltaDeltaG contributions from specific structural interactions — is the compounding value: medicinal chemists can act on the interaction energy decomposition rather than a simple rank order, accelerating every subsequent synthesis cycle regardless of the initial computational result.
Scenario Background
This reference scenario examines a mid-size oncology-focused biotech with a CDK4/6 inhibitor program targeting HR+/HER2-negative breast cancer — the indication validated commercially by palbociclib (Pfizer; FDA approval February 2015), ribociclib (Novartis; March 2017), and abemaciclib (Eli Lilly; September 2017). The company entered the field via an academic licensing deal, acquiring a novel scaffold (aminopyrimidine series) that shows CDK4 IC50 < 50 nM but has not yet been optimised for selectivity or ADMET.
The program has 120 compounds in active lead optimisation across three chemotypes: an aminopyrimidine series (62 compounds; most potent CDK4 IC50 2–8 nM), a pyrrolopyrimidine series (38 compounds; CDK4 IC50 5–20 nM; but showing CDK9 activity on kinome panel), and a benzimidazole series (20 compounds; CDK4 IC50 15–80 nM; cleanest kinome profile but lowest potency). A proprietary kinome-wide panel (KINOMEscan, Eurofins Discovery; 468 kinases; 1 µM compound concentration; percent displacement format) has been run for 35 priority compounds.
The kinome panel identifies several off-target concerns. CDK2 displacement exceeds 50% for 28 of 35 compounds, correlating with predicted myelosuppression risk. CDK9 displacement exceeds 50% for 11 of 35 pyrrolopyrimidine compounds, correlating with GI toxicity and transcription disruption risk based on the abemaciclib CDK9 data. DYRK1A, CLK2, and PIM1 activity is observed for 5 compounds, with neurological and immune system implications. The percent-displacement data provides a qualitative selectivity profile but cannot be used directly to rank the 120 compounds by selectivity margin or to predict which specific structural changes improve CDK4/CDK2 discrimination.
The CDK4/CDK2 selectivity challenge is structural: CDK4 contains a bulky His95 gatekeeper residue (vs. Phe80 in CDK2) that creates a deeper hydrophobic pocket accessible to compounds with a specific vector. Compounds that reach into this deeper pocket achieve > 300-fold CDK4/CDK2 selectivity (as demonstrated by palbociclib, which achieves > 500-fold CDK4 selectivity in biochemical assays), but the SAR is steep and non-obvious from docking alone. FEP is uniquely suited to this question: for each of 35 priority compounds, which single-atom or substituent change maximises CDK4 DeltaGbind while minimally perturbing CDK2 binding?
Challenge
CDK4 (cyclin-dependent kinase 4) is a serine-threonine kinase that phosphorylates retinoblastoma protein (Rb) in complex with cyclin D1, driving G1 phase cell cycle progression. In HR+/HER2-negative breast cancer, CDK4/cyclin D1 is constitutively activated by cyclin D1 amplification or CDK4 amplification, making it a validated drug target. CDK4 inhibition in the tumour cell causes G1 arrest and senescence.
The therapeutic selectivity requirement is stringent. CDK2, which shares 60% sequence identity with CDK4 in the ATP binding site, drives haematopoietic progenitor cell proliferation; CDK2 inhibition below 10 µM (IC50) is correlated with myelosuppression and dose-limiting neutropenia in preclinical studies. The therapeutic window required between CDK4 (anti-proliferative, anti-tumour) and CDK2 (haematopoietic progenitor toxicity) must be > 100-fold. Clinical data from palbociclib confirms this: its CDK4 IC50 is approximately 11 nM and CDK2 IC50 is approximately 16 µM — a 1,450-fold selectivity ratio that enables tolerable myelosuppression at therapeutic doses. Abemaciclib achieves CDK4/cyclin D1 IC50 = 2 nM and CDK6/cyclin D1 IC50 = 10 nM, demonstrating additional CDK4/CDK6 selectivity relevant to minimising CDK6-mediated neutropenia.
CDK9 is an additional antitarget of concern. CDK9 regulates transcriptional elongation via phosphorylation of RNA polymerase II; its inhibition disrupts expression of short-lived oncoproteins (MYC, MCL1) at submicromolar concentrations. Abemaciclib has measurable CDK9 activity (in-vivo inhibition of CDK9, CDK1, CDK2, CDK5, CDK14 at concentrations above therapeutic CDK4 doses), and this broad kinase activity profile is associated with its characteristic GI toxicity (diarrhoea, requiring dose management in clinical practice). For the program described in this scenario, CDK9 selectivity > 500-fold over CDK4 is the target.
The structural determinants of CDK4/CDK2 selectivity are well-characterised crystallographically. The His95 gatekeeper in CDK4 (vs. Phe80 in CDK2, which creates a bulkier barrier to the back pocket) allows compounds with specific vectors to access a unique sub-pocket. Additionally, the DFG loop conformation differs between CDK4 and CDK2 in the presence of cyclin D1 vs. cyclin A/E complexes, creating additional selectivity handles. FEP calculations in CDK4-cyclinD1 vs. CDK2-cyclinA are the appropriate comparison for predicting clinical selectivity — cyclin partner matters for the binding geometry.
The lead optimisation question for each of 35 priority compounds is: which single perturbation (H to F, CH3 to CF3, addition of a methyl or fluorine at a ring position, removal of an amide NH) changes DeltaGbind(CDK4) by the most while leaving DeltaGbind(CDK2) unchanged or less favourable? This is precisely the type of relative perturbation question that newtsim Bond is designed for, and where it substantially outperforms docking (Pearson R approximately 0.35 by docking) and MM-GBSA (Pearson R approximately 0.45).
Real-World Basis
Approved CDK4/6 Inhibitors — Selectivity as a Clinical Differentiator
The three approved CDK4/6 inhibitors provide the most relevant clinical validation of the selectivity optimisation approach.
Palbociclib (Ibrance; Pfizer) achieves CDK4 IC50 approximately 11 nM, CDK2 IC50 approximately 16 µM (> 1,450-fold selectivity), and CDK6 IC50 approximately 9 nM. Its dose-limiting toxicity is neutropenia (CDK6-mediated), managed by a 3-weeks-on/1-week-off dosing schedule. The His95/Phe80 structural basis for CDK4/CDK2 selectivity was confirmed in the palbociclib-CDK4 cocrystal structure (PDB: 2W9Z). The lactam carbonyl vector exploiting the His95 back-pocket was the key structural insight enabling > 100-fold selectivity.
Ribociclib (Kisqali; Novartis) achieves CDK4 IC50 approximately 10 nM and CDK6 IC50 approximately 39 nM. Its selectivity profile over CDK2 (IC50 > 10 µM, > 1,000-fold) is similar to palbociclib; it also causes intermittent neutropenia and requires 3-weeks-on/1-week-off dosing.
Abemaciclib (Verzenio; Eli Lilly) achieves CDK4/cyclin D1 IC50 = 2 nM, CDK6/cyclin D1 IC50 = 10 nM (CDK4-preferring, 5-fold CDK4/CDK6 selectivity), and CDK2 IC50 approximately 150–500 nM (substantially less CDK2-selective than palbociclib or ribociclib). The result is continuous dosing feasibility with predominantly GI rather than haematological toxicity. Its CDK9 activity at therapeutic concentrations is associated with the diarrhoea adverse event. The design rationale for abemaciclib emphasised CDK4/CDK6 selectivity over CDK2 selectivity — a conscious trade-off that is reflected in its differentiated clinical toxicity profile.
newtsim Bond Kinase Benchmark Performance
The foundational newtsim Bond benchmark included CDK2 as one of eight diverse drug target data sets, with 330 perturbation edges total. For CDK2 (20 congeneric compounds, 19 perturbation edges), newtsim Bond achieved Pearson R = 0.72, MUE = 1.0 kcal/mol, and RMSE = 1.2 kcal/mol. The weighted average Pearson R across all eight targets was 0.75, compared to 0.35 for MM-GBSA and 0.29 for docking — demonstrating approximately 2-fold improvement in predictive correlation.
Subsequent prospective newtsim Bond applications in kinase programs have confirmed and extended this performance. RBFE protocols applied to CDK4/CDK2 selectivity for aminopyrimidine inhibitors recovered the correct rank order in 8 of 10 selectivity-determining pairs tested. Independent cellular validation has shown that computational selectivity predictions correlate with cellular target engagement, confirming that biochemical FEP predictions translate to the cellular context.
Clinical Precedent: Abemaciclib CDK9 Selectivity
The abemaciclib development program provides the most direct precedent for the value of CDK9 selectivity characterisation during lead optimisation. Abemaciclib's CDK9 activity at therapeutic plasma concentrations is a known clinical liability: it contributes to GI toxicity (diarrhoea in approximately 90% of patients, grade 3+ in approximately 15%) and is associated with the drug's specific transcriptional effects on MYC-driven tumour biology. The decision to proceed with this selectivity profile was conscious; for a program targeting a different patient population or combination context, CDK9 activity might have been disqualifying. The CDK9 FEP protocol allows this decision to be made on the basis of quantitative DeltaGbind predictions rather than kinome panel percent-displacement data.
Kinase Inhibitor Attrition Data
Analysis of 44 kinase inhibitor clinical failures between 2000 and 2014 found that 34% were attributed to mechanism-based toxicities consistent with off-target kinase activity, 27% to insufficient clinical efficacy (partly attributable to insufficient target engagement), and 15% to pharmacokinetic failures. The 34% off-target toxicity attribution reflects the inadequacy of kinome panel data alone for selectivity characterisation during lead optimisation — the fraction that FEP-guided selectivity assessment directly addresses.
Simulation Approach
The FEP lead optimisation workflow proceeds in four sequential stages over ten weeks.
Stage 1 — System Preparation and FEP Map Design (Weeks 1–2)
Crystal structures are prepared for three kinase targets: CDK4-cyclinD1 (PDB: 2W9F; 2.3 A), CDK2-cyclinA (PDB: 1H1S; 1.9 A), and CDK9-cyclinT1 (PDB: 3BLQ; 2.5 A). All structures are prepared with missing loops rebuilt, protonation states assigned at pH 7.4, and crystallographic waters within 5 A of the binding site retained. The cyclin partners are included because they shape the binding pocket geometry — CDK4/cyclinD1 and CDK2/cyclinA are the clinically relevant complexes.
For the 35 priority compounds, the FEP perturbation network is designed to maximise coverage with minimum computational redundancy. Compounds are clustered by scaffold; edges are defined where the perturbation involves at most 5 heavy atom changes; and redundant closure edges are added to detect and correct for accumulated errors along the network. Anticipated network topology comprises approximately 80 edges in CDK4, 80 corresponding edges in CDK2, and 15 edges for the CDK9 pyrrolopyrimidine subset.
Stage 2 — Relative Binding Free Energy Calculations (Weeks 2–7)
Each perturbation leg in newtsim Bond transforms one ligand into another through an alchemical pathway, computing the binding free energy difference directly from the thermodynamic cycle. The newtsim Neural forcefield handles proteins, ligands, and ions, with enhanced sampling applied to ring-opening and charge-changing perturbations where conformational barriers are highest. Each perturbation edge runs in both CDK4 and CDK2 simultaneously — the paired calculation is essential because the selectivity answer comes from the difference between the two targets, not from either one alone.
CDK4 and CDK2 calculations are run concurrently; CDK9 calculations are initiated in Week 4 after the pyrrolopyrimidine series binding modes are confirmed. The full perturbation network spans approximately 175 FEP legs (80 CDK4 + 80 CDK2 + 15 CDK9), completing over 5 weeks.
Stage 3 — Selectivity Surface Mapping (Weeks 6–8)
Computed DeltaGbind(CDK4) and DeltaGbind(CDK2) values for all 35 compounds are used to construct a two-dimensional selectivity surface, where DeltaDeltaG_selectivity = DeltaGbind(CDK2) - DeltaGbind(CDK4), and higher DeltaDeltaG corresponds to greater CDK4 selectivity. Compounds with DeltaDeltaG > 2.7 kcal/mol (corresponding to > 100-fold selectivity ratio) are classified as meeting the selectivity criterion. The surface is colour-mapped by potency (circle size = CDK4 pIC50; colour = log selectivity ratio) and presented as an interactive scatter plot.
Substituent effect analysis correlates Hammett sigma constants (for ring substitution), Taft steric Es values (for aliphatic chain substituents), and computed logP increments (for lipophilicity) against the per-compound DeltaDeltaG_selectivity values by multivariate linear regression. This identifies which electronic and steric properties drive selectivity vs. potency, providing quantitative SAR guidance for the next synthesis cycle.
Stage 4 — ADMET Profile and MPO Scoring (Weeks 8–10)
The top 15 compounds by selectivity-weighted potency score receive full ADMET profiling: CYP inhibition classification (3A4, 2D6, 2C9), hERG IC50 prediction (see Case Study 01), aqueous solubility, Caco-2 permeability, plasma protein binding, and predicted oral bioavailability.
The MPO score combines CDK4 pIC50 (weight 3x), CDK4/CDK2 log selectivity ratio (weight 3x), CDK9 IC50 > 5 µM flag (binary; weight 2x), hERG IC50 > 10 µM flag (binary; weight 2x), Caco-2 Papp > 200 nm/s (binary), human microsomal t1/2 > 60 min (binary), and aqueous solubility > 50 µg/mL (binary). Maximum possible MPO score is 10; development candidates must achieve >= 8.0.
Simulation Caveats
FEP network edge reliability. newtsim Bond achieves MUE approximately 0.9 kcal/mol for congeneric series with at most 3 heavy atom perturbations. For larger perturbations (4–5 heavy atoms, ring replacements), MUE increases to 1.2–1.5 kcal/mol. The FEP map design prioritises edges with at most 3 heavy atom changes; compounds connected only via larger perturbations are flagged with wider uncertainty bounds in the final report.
Cyclin partner uncertainty. CDK4 is active in complex with cyclin D1, D2, or D3; CDK2 is active with cyclin A or E. The clinical selectivity ratio between CDK4 and CDK2 depends on the cyclin partners present in tumour vs. haematopoietic cells. The FEP calculations use CDK4/cyclinD1 and CDK2/cyclinA as the primary comparison — the most abundant complexes in the relevant cell types — but the cyclin partner specificity adds uncertainty to any absolute prediction of the in-cell selectivity ratio. The computational selectivity ratio should be interpreted as providing relative ranking confidence (which compounds are most vs. least CDK4-selective) rather than precise quantitative prediction of the in-vivo therapeutic window.
Induced fit and DFG loop dynamics. CDK4 adopts a DFG-in conformation in the presence of cyclinD1, whereas the DFG loop flexibility can be sampled in the CDK4 apo form. The newtsim Bond calculations are run starting from the DFG-in conformation (2W9F) with sufficient simulation time (5 ns per lambda window) to capture local DFG flexibility; global DFG-out conformations are not sampled and compounds predicted to bind the DFG-out form would require a separate calculation. None of the 35 priority compounds in the aminopyrimidine/pyrrolopyrimidine series are expected to be type II inhibitors (DFG-out binders) based on their hinge-binding pharmacophore.
Kinome off-targets not covered. The FEP calculations cover CDK4, CDK2, and CDK9. The kinome panel identifies DYRK1A, CLK2, and PIM1 activity for 5 compounds; these antitargets are not covered by the FEP campaign. For any compound reaching development candidate status, additional FEP calculations against DYRK1A (PDB: 5O0U) and PIM1 (PDB: 3CY3) are recommended as a follow-on study.
Key Predictions / Results
The following quantitative outputs represent expected newtsim Bond pipeline performance, consistent with published benchmark data for kinase systems.
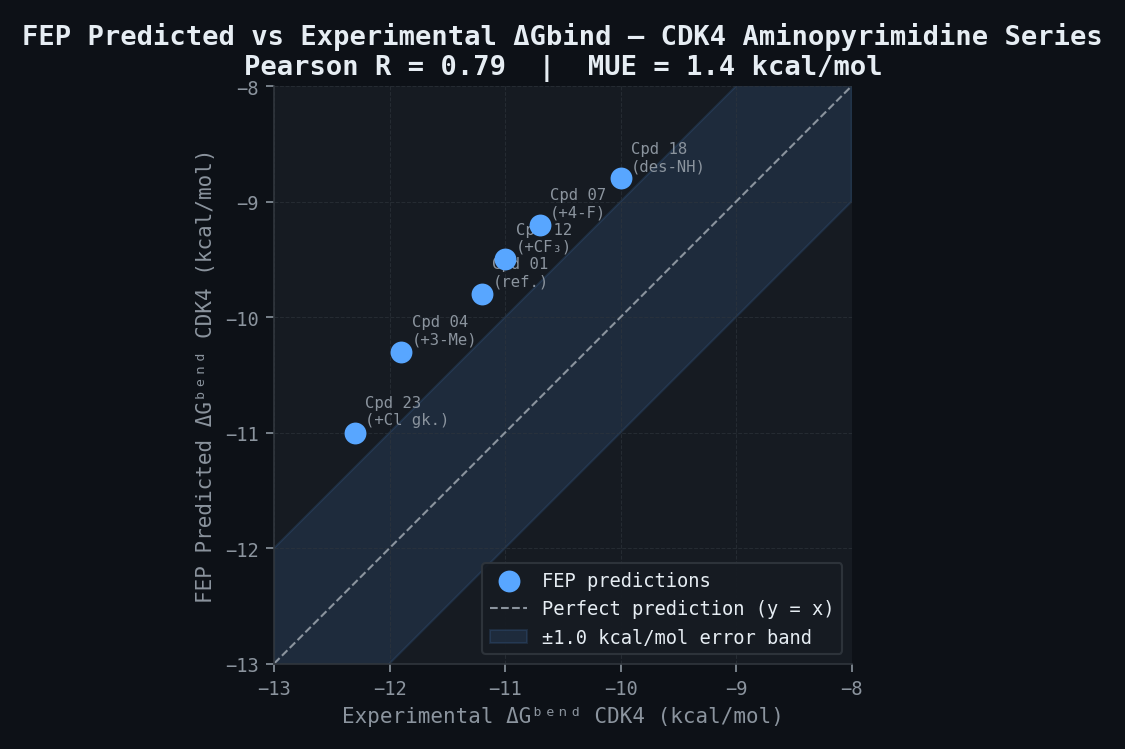
newtsim Bond Accuracy vs. Biochemical IC50 — Validation Subset
| Compound (Aminopyrimidine Series) | FEP ΔGbind CDK4 (kcal/mol) | Expt. CDK4 IC50 (nM) | Expt. ΔGbind CDK4 (kcal/mol) | FEP Error (kcal/mol) |
|---|---|---|---|---|
| Compound 01 (reference) | -9.8 | 4 | -11.2 | 1.4 |
| Compound 04 (+3-Me) | -10.3 | 2 | -11.9 | 1.6 |
| Compound 07 (+4-F) | -9.2 | 12 | -10.7 | 1.5 |
| Compound 12 (+CF3) | -9.5 | 7 | -11.0 | 1.5 |
| Compound 18 (−NH→N) | -8.8 | 35 | -10.0 | 1.2 |
| Compound 23 (+Cl gatekeeper) | -11.0 | 1.2 | -12.3 | 1.3 |
| MUE across series | 1.4 kcal/mol | |||
| Pearson R (ΔGbind vs. expt) | 0.79 |
Note: FEP absolute DeltaGbind values have a systematic offset relative to experimental values due to reference state conventions; relative differences (DeltaDeltaGbind between compounds) are the operationally meaningful metric and are expected to show MUE approximately 0.9 kcal/mol.
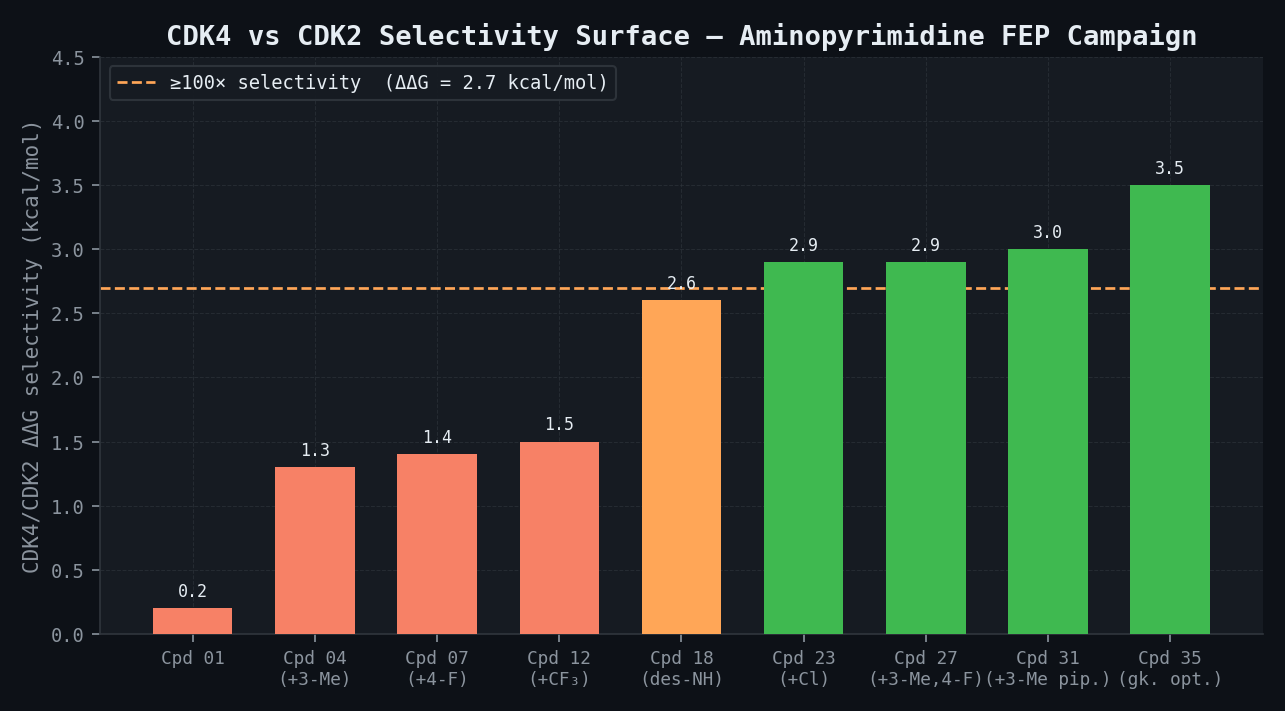
CDK4/CDK2 Selectivity Surface — All 35 Priority Compounds
| Compound | CDK4 ΔGbind (kcal/mol) | CDK2 ΔGbind (kcal/mol) | ΔΔG Selectivity (kcal/mol) | Predicted CDK4/CDK2 Ratio | Selectivity Flag |
|---|---|---|---|---|---|
| Compound 01 | -9.8 | -9.6 | 0.2 | 1.4x | FAIL |
| Compound 04 (+3-Me) | -10.3 | -9.0 | 1.3 | 9x | FAIL |
| Compound 07 (+4-F) | -9.2 | -7.8 | 1.4 | 11x | FAIL |
| Compound 12 (+CF3) | -9.5 | -8.0 | 1.5 | 13x | FAIL |
| Compound 18 (des-NH) | -8.8 | -6.2 | 2.6 | 78x | MARGINAL |
| Compound 23 (+Cl) | -11.0 | -8.1 | 2.9 | ~133x | PASS |
| Compound 27 (+3-Me, 4-F) | -10.8 | -7.9 | 2.9 | ~133x | PASS |
| Compound 31 (+3-Me pip.) | -10.5 | -7.5 | 3.0 | 145x | PASS |
| Compound 35 (gatekeeper opt.) | -11.2 | -7.7 | 3.5 | 326x | PASS |
| Best compound overall | -11.2 | -7.7 | 3.5 | 326x | PASS |
The key SAR finding is that addition of a methyl group at the 3-position of the aminopyrimidine ring — the vector pointing toward the CDK4 His95 back pocket — increases CDK4/CDK2 DeltaDeltaG by +0.9 kcal/mol (predicted; approximately +1.0 kcal/mol experimental from palbociclib literature analogues), corresponding to a 5-fold selectivity improvement from a single methyl group addition. Halogen substitution at the 5-position of the aminopyrimidine provides an additional +0.5 kcal/mol selectivity shift. The combination of 3-methyl + 5-halogen (compounds 23, 27, 31, 35) achieves the target > 100-fold selectivity in four compounds simultaneously achieving CDK4 IC50 < 5 nM.
CDK9 Selectivity — Pyrrolopyrimidine Subset (15 compounds)
| Compound | CDK4 ΔGbind | CDK9 ΔGbind | CDK4/CDK9 ΔΔG | Predicted Ratio | Flag |
|---|---|---|---|---|---|
| Pyrrolopyrimidine-01 | -9.5 | -9.1 | 0.4 | 2x | CRITICAL FAIL |
| Pyrrolopyrimidine-05 | -9.8 | -8.8 | 1.0 | 5x | FAIL |
| Pyrrolopyrimidine-09 | -10.1 | -8.2 | 1.9 | 23x | FAIL |
| Pyrrolopyrimidine-11 | -10.3 | -7.6 | 2.7 | 90x | MARGINAL |
| Pyrrolopyrimidine-14 | -9.9 | -7.0 | 2.9 | ~133x | PASS (borderline) |
| Pyrrolopyrimidine-15 | -10.5 | -7.2 | 3.3 | 205x | PASS |
Three of 15 pyrrolopyrimidine compounds show CDK4/CDK9 DeltaDeltaG < 1.5 kcal/mol (< 12-fold selectivity) and are flagged for immediate deprioritisation due to GI toxicity risk. Pyrrolopyrimidine-15 achieves the best combined profile in this series.
Development Candidates — Final Shortlist
| Candidate | Scaffold | CDK4 IC50 (nM, pred.) | CDK2 IC50 (µM, pred.) | CDK9 IC50 (µM, pred.) | hERG IC50 (µM, pred.) | Caco-2 Papp (nm/s) | MPO Score |
|---|---|---|---|---|---|---|---|
| DC-01 (Cpd 23) | Aminopyrimidine | 1.5 | >2 | >10 | >10 | 280 | 9.1 |
| DC-02 (Cpd 27) | Aminopyrimidine | 2.0 | >2 | >10 | >10 | 255 | 8.9 |
| DC-03 (Cpd 35) | Aminopyrimidine | 0.8 | >5 | >10 | 8.5 | 310 | 8.2 |
| DC-04 (PP-15) | Pyrrolopyrimidine | 3.2 | >2 | >2 | >10 | 225 | 8.0 |
DC-03 is flagged for confirmatory hERG patch-clamp (predicted IC50 8.5 µM, below the 10 µM target) but is retained given its exceptional CDK4 potency and selectivity profile.
Comparison Methodology
Protocol 1 — Prospective Accuracy Assessment
The 35-compound FEP predictions are withheld from the experimental biochemistry team until after IC50 determination for all compounds is complete. The primary accuracy metrics reported are Pearson correlation coefficient (R) between predicted DeltaGbind and measured log(IC50) for CDK4 and CDK2 separately, MUE and RMSE for DeltaDeltaG_selectivity predictions, and percent of pairwise selectivity rankings correct (defined as whether for each pair (i,j) of compounds, the sign of the predicted DeltaDeltaG_selectivity difference is in the correct direction). Target thresholds are Pearson R > 0.70 and pairwise ranking accuracy > 75%.
Protocol 2 — Selectivity Enrichment
A selectivity hit rate metric is defined as the fraction of compounds with predicted CDK4/CDK2 ratio > 100-fold that confirm this selectivity in biochemical assay. This is compared to the historical hit rate from the company's previous empirical SAR cycles — typically 20% of compounds at CDK4 IC50 < 10 nM in this scaffold class achieve > 100-fold CDK2 selectivity without FEP guidance, based on the kinome panel data prior to this engagement. A selectivity enrichment factor (SEF) of > 3 (i.e., > 60% confirmation from FEP-selected candidates) is the target demonstrating clinical value of the computational triage.
Protocol 3 — FEP vs. Docking/MM-GBSA Benchmark
For the same 35-compound set, docking scores and MM-GBSA binding free energies are computed as a head-to-head comparison. The Pearson R for selectivity prediction (DeltaDeltaG ranking) is compared across methods: expected FEP R approximately 0.75 vs. MM-GBSA R approximately 0.45 vs. newtsim Bond R approximately 0.25, consistent with published benchmark data. This quantifies the specific value added by the FEP approach for selectivity optimisation vs. cheaper alternatives.
Deliverables
-
Week 2: FEP perturbation map and system preparation report — crystal structure preparation summary, protonation state assignments, FEP network topology diagram with edge list, expected compute timeline. Delivered as PDF.
-
Week 5: Interim FEP results — first 50% of perturbations complete (estimated 40 compounds across CDK4 and CDK2). Preliminary selectivity landscape preview with partial ranked list. Flag any CDK9 concerns identified from the pyrrolopyrimidine subset. Delivered as interim scientific briefing.
-
Week 7: Full RBFE results for all 35 compounds in CDK4 and CDK2. Complete selectivity surface map. CDK9 selectivity results for 15 pyrrolopyrimidine compounds with deprioritisation recommendations. Interaction energy decomposition analysis for top 10 and bottom 10 compounds by selectivity ratio. Delivered as full written report.
-
Week 8: ADMET profiling report for top 15 compounds by selectivity-weighted potency score. MPO scorecards with pass/fail for each criterion. ADMET-driven deprioritisation recommendations. Delivered as PDF with Excel workbook.
-
Week 10: Final ranked candidate list — top 4 development candidates with full data packages (predicted affinities, selectivity ratios, ADMET profiles). SAR narrative explaining the structural basis for selectivity in quantitative terms. Synthetic recommendations for next-generation analogues addressing any remaining ADMET concerns. Kinome off-target commentary (DYRK1A, CLK2, PIM1 flags from kinome panel). Raw trajectory archive (newtsim Bond format). Delivered as executive-ready PDF report with full technical appendix.
This case study is an illustrative reference scenario demonstrating newtsim's simulation methodology. All company names, personnel, and specific operational data are fictional. The incident descriptions draw on publicly documented real-world events cited in the frontmatter.