Catching Reactive Metabolite Formation Early: The Troglitazone Hepatotoxicity Blueprint
Executive Summary
On 21 March 2000, the FDA withdrew its approval of Rezulin (troglitazone), a first-in-class thiazolidinedione PPARgamma agonist that had been prescribed to hundreds of thousands of patients with type 2 diabetes. At the time of withdrawal, at least 63 deaths from fulminant hepatic failure had been confirmed, along with hundreds of cases of severe liver injury requiring hospitalisation or transplantation. The drug had been on the market for just 38 months — approved by the FDA in January 1997 and generating cumulative sales exceeding $2.1 billion for Warner-Lambert before it was gone. The hepatotoxicity was idiosyncratic in incidence — approximately 1 in 50,000–100,000 patients progressed to clinically significant liver injury, and 1 in 100,000 to fatal liver failure — making it statistically invisible in the clinical trial populations that had cleared the drug for approval. That invisibility was not the same as unpredictability. The molecular mechanism that killed those patients was written in the compound's structure.
CYP3A4-mediated oxidation of troglitazone's chroman ring generates an electrophilic ortho-quinone methide intermediate that covalently alkylates hepatic proteins — a direct initiating event for idiosyncratic hepatotoxicity. A parallel mechanism, also linked to the chromanol moiety, uncouples mitochondrial electron transport, depleting hepatocyte ATP and compounding cell death. The chroman ring is a well-characterised Tier 1 structural alert for reactive metabolite formation. GSH trapping assays, which were not standard practice in the mid-1990s IND-enabling package, would have detected the adducts. The structural flag was present and accessible. The proof came retrospectively: rosiglitazone (Avandia, GSK, approved 1999) and pioglitazone (Actos, Takeda, approved 1999) both lack the chroman ring entirely. Neither compound generates detectable GSH adducts at pharmacologically relevant concentrations. Their hepatotoxicity signal is dramatically lower. The chroman was the problem, and eliminating it was the solution — a solution that arrived only after the deaths.
Had a QM/MM reactive metabolite pipeline been deployed during lead optimisation, the chroman liability would have been identified before IND submission. The key mechanism: CYP3A4 oxidation at C-5 of the chroman ring generates an ortho-quinone methide with a GSH adduct activation energy (DeltaG-dagger) of approximately 12.8 kcal/mol — well below the 15 kcal/mol HIGH risk threshold — and a computed spin density of 0.48 on the exocyclic carbon, confirming facile Michael addition to protein nucleophiles. All 22 chroman-TZD compounds in a representative 85-compound PPARgamma library would be classified HIGH reactive metabolite risk. FEP-guided scaffold hopping would then generate chroman-free bioisosteres — specifically a 3-methylpyridine replacement (DeltaDeltaGbind vs. troglitazone: +0.3 kcal/mol; reactive metabolite risk: LOW) — pointing directly toward the safer chemotypes that became rosiglitazone and pioglitazone.
Had this simulation been run during lead optimisation, all 22 chroman-TZD compounds would have been classified HIGH reactive metabolite risk before any IND-enabling toxicology studies, and the bioisostere library would have pointed directly toward the safer chemotypes that became rosiglitazone and pioglitazone. Rezulin was withdrawn on 21 March 2000 after at least 63 confirmed deaths from fulminant hepatic failure, with cumulative sales of 136 million at the time of Pfizer's acquisition — with true liability costs estimated to have substantially exceeded this. The diabetes development field sustained broader damage as the entire TZD class faced sustained FDA scrutiny and rosiglitazone was later restricted on cardiovascular grounds — a cascade rooted in a chroman substituent that computational reactive metabolite screening would have flagged before first patient exposure. Where long-term metabolite monitoring is relevant — particularly for compounds advancing through IND-enabling studies — newtsim livesim provides continuous integration of emerging CYP inhibition and GSH trapping data against the computational liability predictions, updating risk classifications in real time.
Scenario Background
This reference scenario examines a large pharmaceutical company with an active metabolic disease franchise, advancing three PPARgamma agonist development candidates toward IND-enabling toxicology studies in the mid-1990s. The program has a compound library of 85 thiazolidinedione and non-TZD PPARgamma agonists at varying stages of optimisation, covering four structural chemotypes: chroman-TZDs (the troglitazone class), benzofuran-TZDs, indanone-TZDs, and non-TZD PPARgamma partial agonists. Approximately 30% of the library — approximately 25 compounds — contain chroman, benzofuran, or substituted indanone scaffolds known to be CYP oxidation substrates with reactive metabolite risk.
The program's standard ADMET panel at this stage (mid-1990s) includes microsomal stability in rat and human liver microsomes (substrate depletion assay; t1/2 measurement), Caco-2 bidirectional permeability, plasma protein binding (ultracentrifugation), and CYP inhibition panel (3A4, 2C9, 2D6 at 10 µM). None of these assays directly detect covalent protein adduct formation, reactive metabolite trapping, or mitochondrial toxicity. GSH trapping assays — in which reactive electrophilic metabolites are captured by reduced glutathione and detected by LC-MS/MS — were not standard practice in the pharmaceutical industry until after the troglitazone withdrawal. The reactive metabolite safety assessment landscape of 1995–1997 was, in retrospect, substantially inadequate.
The program timeline places compounds 12–18 months from anticipated IND submission. Three chroman-TZDs show PPARgamma IC50 < 50 nM in a TR-FRET displacement assay and full agonist activity in 3T3-L1 adipocyte differentiation — the same profile as troglitazone. Two non-TZD partial agonists show lower PPARgamma efficacy but better selectivity over PPARalpha (important for avoiding cardiac side effects). The simulation engagement is structured as: (1) reactive metabolite risk screen across all 85 compounds; (2) QM/MM characterisation of flagged compounds; (3) FEP-guided scaffold hopping to generate chroman-free analogues preserving PPARgamma binding; and (4) integrated ADMET profiling for the retained set.
Challenge
PPARgamma (peroxisome proliferator-activated receptor gamma) is a nuclear receptor with a large, flexible ligand-binding domain (LBD volume approximately 1,300 A³) that accommodates a wide structural diversity of lipophilic agonists. The LBD cavity can be divided into two arms: the activation function 2 (AF-2) helix-binding arm, which is occupied by the TZD or other acidic head group that contacts His323 and Tyr473 in the AF-2 activation helix, and the hydrophobic extension arm, which accommodates the lipophilic tail. In the TZD series, the chroman ring fills the hydrophobic arm and provides significant binding surface — making it difficult to substitute without sacrificing PPARgamma affinity.
The simulation challenge has two critical components. The first is reactive metabolite prediction: identifying which compounds in the 85-compound library undergo CYP-mediated bioactivation to electrophilic intermediates capable of covalently modifying hepatic proteins. The chroman ring undergoes CYP3A4-mediated oxidation at the C-5 position (adjacent to the phenolic OH) to generate an ortho-quinone methide — a potent, short-lived Michael acceptor that reacts efficiently with cysteine and lysine nucleophiles in GSH and hepatic proteins. This mechanism was demonstrated definitively using isotope labelling and LC-MS/MS adduct characterisation; the reactive metabolite formed with methanethiol (a GSH mimic) with an activation energy consistent with rapid, nonenzymatic Michael addition.
The second component is mitochondrial toxicity. Subsequent work demonstrated that troglitazone and several chroman-TZD analogues uncouple the mitochondrial electron transport chain by dissipating the proton gradient, leading to ATP depletion, mitochondrial membrane depolarisation, and hepatocyte energy crisis. This mechanism is parallel to — and additive with — the reactive metabolite covalent binding. The mitochondrial liability is more difficult to predict computationally and is assessed semi-quantitatively in the pipeline via lipophilicity and membrane partitioning descriptors correlated against the known mitochondriotoxic potency of troglitazone and analogues.
The third challenge is selectivity. PPARgamma partial agonists — which show lower transcriptional efficacy but retain sufficient insulin-sensitising activity — avoid the full agonist-associated adverse effects (weight gain, fluid retention, cardiac effects) while maintaining therapeutic benefit. Identification of the structural features that confer partial agonism, and their compatibility with the chroman-free scaffold hop, requires explicit MD simulation of the AF-2 helix dynamics in complex with each scaffold type.
Real-World Basis
The troglitazone withdrawal is among the most consequential drug safety events in modern pharmacology, with mechanistic, regulatory, and commercial implications that extend far beyond the TZD class.
Regulatory Timeline and Epidemiology
Parke-Davis/Warner-Lambert submitted troglitazone for FDA review on 31 July 1996. FDA approved Rezulin on 29 January 1997, and the drug appeared in pharmacies in late March 1997. By July 1997, Rezulin had captured 12% of the US oral diabetes market; at 2.1 billion by the time of withdrawal.
Hepatotoxicity signals emerged rapidly post-approval. In December 1997, Glaxo Wellcome withdrew troglitazone from the UK market after hepatotoxicity concerns. Japan withdrew the drug in 2000. In the United States, FDA convened advisory panels in 1998 and 1999; the agency mandated increasingly frequent liver function test monitoring but resisted withdrawal under pressure from Warner-Lambert, which argued that the drug's glycaemic control benefits outweighed its risks for a population with few alternatives. The pivotal event was the FDA's own Office of Drug Safety reporting 94 cases of acute liver failure by early 2000 — 89 acute and 5 chronic — with 58 cases (67%) in women, and only 11 of the 89 acute cases (13%) recovering without liver transplantation. On 21 March 2000, FDA withdrew Rezulin's approval. By this time, rosiglitazone and pioglitazone — both lacking the chroman ring and thus the primary reactive metabolite mechanism — had been approved and were capturing the TZD market.
Mechanistic Characterisation
CYP3A4 and CYP2C8 oxidise the chroman ring at the C-5 position (the carbon ortho to the phenolic OH) to generate a reactive ortho-quinone methide intermediate. This electrophile reacts with the thiol of glutathione (and hepatic protein cysteines) to form stable Michael adducts detectable by LC-MS/MS. The formation of quinone and ortho-quinone methide reactive metabolites occurs by two parallel pathways: oxidation of the OH group (generating the quinone) and oxidation of the methyl groups ortho to the OH (generating the quinone methide). Both pathways generate electrophilic species reactive toward biological nucleophiles. This mechanism was definitively confirmed using isotope labelling and LC-MS/MS adduct characterisation.
A parallel hepatotoxic mechanism involves mitochondrial uncoupling: troglitazone and its metabolites partition into the mitochondrial inner membrane and uncouple the proton gradient, causing membrane potential collapse and ATP depletion. The mitochondrial uncoupling IC50 of troglitazone in isolated rat liver mitochondria is approximately 5–10 µM; the toxic plasma concentrations in susceptible patients may approach this range due to CYP induction (troglitazone is a potent CYP3A4 inducer, potentially elevating its own plasma concentrations over time).
Structural Contrast: Rosiglitazone and Pioglitazone
Rosiglitazone (Avandia) and pioglitazone (Actos), both approved in 1999, lack the chroman ring entirely. Rosiglitazone substitutes a pyridyl-methyl group for the chroman; pioglitazone uses a pyridyl-ethyl group. Neither compound generates detectable GSH adducts in standard LC-MS/MS trapping assays at pharmacologically relevant concentrations. Their clinical hepatotoxicity signal is dramatically lower than troglitazone — though not absent, as idiosyncratic liver injury is a class effect with any metabolically activated compound, and rosiglitazone has its own cardiovascular controversy. The structural lesson — chroman elimination eliminates the primary reactive metabolite risk — was clear in retrospect and is reproducible prospectively by the QM/MM pipeline described here.
Computational Precedent
QM/MM studies of CYP3A4 oxidation of aromatic systems demonstrate that the regioselectivity of oxidation can be predicted from spin density on the substrate radical intermediate, accessible via DFT calculations. Rule-based and ML models trained on known reactive metabolites achieve > 75% sensitivity for electrophilic metabolite risk at the scaffold level. The chroman scaffold is a Tier 1 structural alert — it would be flagged in the first stage of any properly constructed reactive metabolite screen.
Simulation Approach
The metabolic activation pipeline for this 85-compound PPARgamma agonist program proceeds in five stages over nine weeks.
Stage 1 — Structural Alert Screening and Metabolic Site Prediction (Week 1)
All 85 compounds are processed through a comprehensive structural alert filter covering chroman rings, benzofurans, anilines, hydroquinones, catechols, alpha,beta-unsaturated carbonyls, Michael acceptors, epoxide-forming alkenes, and thiophenes. Compounds passing primary alert screening proceed directly to ADMET profiling. Compounds triggering alerts proceed to metabolic site of metabolism (SOM) prediction using a regioselectivity model trained on CYP3A4 and CYP2C8 substrates (> 800 compounds). SOMs are ranked by predicted oxidation likelihood; sites with probability > 0.4 are designated primary SOMs for QM/MM treatment. The purpose of this stage is rapid triage: approximately half the library is expected to clear the alert screen, concentrating the expensive QM/MM computation on the compounds that need it.
Stage 2 — QM/MM Reactive Metabolite Characterisation (Weeks 2–5)
For approximately 25 compounds flagged with a primary alert and a CYP-susceptible SOM prediction, a QM/MM protocol models the oxidation event directly. The CYP3A4 active site is modelled using the substrate-bound crystal structure (PDB: 1TQN; 2.15 A). Each compound is docked into the active site in the oxidation-ready geometry — substrate positioned within 3.5 A of the heme iron at the predicted SOM — using a constrained newtsim Bond protocol. The docked complex is partitioned for QM/MM: the compound plus the heme group (~70 atoms) form the quantum region, computed with newtsim Root; the surrounding protein environment forms the classical region.
The oxidised substrate radical intermediate is generated by abstracting a hydrogen atom from the predicted SOM using the Compound I CYP active species (FeIV=O porphyrin cation radical). The spin density distribution on the oxidised intermediate determines whether the intermediate is electrophilic: compounds where the spin density on the oxidised carbon predicts stabilisation of an ortho-quinone methide (spin density > 0.3 on the exocyclic carbon) or an arene oxide are scored as HIGH reactive metabolite risk. The rationale for the QM/MM approach rather than a simpler rule-based method is that spin density quantifies the electrophilic character of the actual intermediate — it distinguishes genuinely dangerous quinone methides from benign N-oxides that trigger the same structural alerts.
Stage 3 — Covalent Adduct Energetics (Weeks 4–6)
For confirmed high-risk compounds (predicted quinone methide or epoxide intermediate), the activation energy for GSH adduct formation is computed. The electrophilic intermediate reacts with methanethiol (CH3SH; a standard GSH cysteine mimic) in a quantum chemical model using newtsim Root. The transition state is located and the activation free energy DeltaG-dagger is computed with thermal and entropic corrections at 298 K. A threshold of DeltaG-dagger < 15 kcal/mol corresponds to HIGH covalent binding risk, calibrated against published benchmarks for known GSH-reactive electrophiles. This stage answers a specific question: given that the intermediate is electrophilic, how readily does it react with biological nucleophiles? Compounds below the threshold react fast enough to produce significant covalent protein adducts at pharmacologically relevant concentrations.
Stage 4 — Scaffold Hopping with Binding Mode Conservation (Weeks 5–8)
For each HIGH-risk compound, a bioisostere library is generated by replacing the reactive moiety with fragments that eliminate the structural alert. For the chroman scaffold, proposed bioisosteric replacements include 3-methylpyridine (the rosiglitazone approach), cyclopentyl, 3-fluorophenyl, pyridone, and tetrahydropyran. For benzofuranyl scaffolds, replacements include indanyl or methyl-substituted phenyl.
The PPARgamma binding free energy of each bioisostere is computed using the newtsim Bond relative binding free energy (RBFE) protocol, using the troglitazone PPARgamma cocrystal structure (PDB: 2PRG; 2.2 A) as the reference. Each perturbation map connects the high-risk parent compound to its 2–4 best bioisostere candidates via small structural perturbations (at most 5 heavy atoms changed per edge), computed with newtsim Neural forcefield. The objective criteria for a retained bioisostere are: (a) reactive metabolite alert eliminated, (b) PPARgamma DeltaDeltaGbind within +/- 1.0 kcal/mol of parent, and (c) PPARgamma/PPARalpha selectivity ratio > 50-fold preserved. The rationale for FEP rather than docking at this stage is that scaffold hops change ring systems, which alters the shape of the binding pocket occupancy in ways that docking scores do not reliably capture — FEP propagates these changes through the full thermodynamic cycle.
Stage 5 — Integrated ADMET Profiling (Weeks 7–9)
All retained compounds — original non-flagged (~49) plus scaffold-hopped bioisosteres (~30) — receive full ADMET profiling: computed logP (newtsim Neural electrostatics), logD7.4, aqueous solubility, Caco-2 permeability, plasma protein binding, CYP inhibition risk assessment (2D6, 3A4, 2C9), and hERG IC50 prediction (see Case Study 01 methodology). A multi-parameter optimisation (MPO) score is computed combining potency (PPARgamma pIC50 >= 8), selectivity (PPARgamma/PPARalpha ratio >= 50), solubility (logS >= -4), metabolic stability (human microsomal t1/2 >= 60 min), reactive metabolite risk (LOW only), and hERG IC50 (>= 10 µM). The top 10 compounds by MPO score are ranked with synthetic accessibility assessment.
Simulation Caveats
Idiosyncratic toxicity vs. direct toxicity. Troglitazone's hepatotoxicity is idiosyncratic — its incidence of approximately 1 in 50,000–100,000 is far below any clinical trial's statistical power to detect. The reactive metabolite mechanism explains the molecular initiating event (covalent protein binding) but does not predict which individuals are susceptible. Susceptibility factors likely include individual variation in CYP3A4 activity, NAT2 genotype, immune response predisposition, and hepatic GSH availability. The simulation pipeline predicts reactive metabolite risk at the scaffold level — which is the actionable information for compound selection — but cannot predict individual patient susceptibility. The correct use of this output is to eliminate HIGH-risk scaffolds from the development pipeline, not to predict incidence in any defined patient population.
QM/MM model limitations. The B3LYP/6-31G* level of theory provides qualitatively reliable spin density predictions and semiquantitative activation energy estimates, but absolute DeltaG-dagger values carry +/- 2–3 kcal/mol uncertainty relative to higher-level DLPNO-CCSD(T) or PBE0-D3 benchmarks. The 15 kcal/mol threshold is an empirically calibrated value — compounds below this threshold in the literature have > 70% GSH trapping positivity; compounds above it have < 30%. The classification is reliable for HIGH vs. LOW decisions; the MEDIUM zone (DeltaG-dagger 12–17 kcal/mol) requires in vitro GSH trapping confirmation.
Mitochondrial toxicity prediction. The mitochondrial uncoupling mechanism of troglitazone is not fully captured by the reactive metabolite pipeline and is assessed only semi-quantitatively via lipophilicity (logP) and computed membrane partitioning. Compounds with logP > 4.5 and a phenol or chromanol moiety are flagged for mitochondrial liability consideration; in vitro Seahorse XF assay confirmation is recommended for any compound proceeding to IND.
FEP accuracy for scaffold-hopped bioisosteres. The newtsim Bond protocol achieves MUE approximately 0.8–1.0 kcal/mol for congeneric series (published benchmark data). For scaffold-hopped bioisosteres — where the perturbation changes ring systems rather than substituents — accuracy may degrade to MUE 1.2–1.5 kcal/mol due to increased alchemical path complexity. The +/- 1.0 kcal/mol acceptance criterion for bioisostere binding affinity should be interpreted with this caveat; experimental TR-FRET confirmation is recommended for all selected bioisosteres before progression.
Key Predictions / Results
The following quantitative outputs represent the expected results from this illustrative pipeline for this program, anchored to published benchmark performance.
Library-Wide Reactive Metabolite Risk Classification
| Chemotype | Compounds in Library | HIGH Risk (n) | MEDIUM Risk (n) | LOW Risk (n) | Primary Alert |
|---|---|---|---|---|---|
| Chroman-TZDs | 22 | 18 | 3 | 1 | Chroman quinone methide |
| Benzofuran-TZDs | 8 | 4 | 2 | 2 | Benzofuran epoxide |
| Indanone-TZDs | 6 | 0 | 4 | 2 | Indanone enone |
| Non-TZD PPARγ agonists | 49 | 0 | 5 | 44 | Various minor alerts |
| Total (85 compounds) | 85 | 22 | 14 | 49 |
QM/MM Reactive Metabolite Characterisation — Troglitazone vs. Rosiglitazone Reference
| Compound | CYP Isoform | Primary SOM | Predicted Intermediate | Spin Density (exo-C) | GSH Adduct ΔG‡ (kcal/mol) | Risk Classification |
|---|---|---|---|---|---|---|
| Troglitazone | CYP3A4 | C-5 chroman | ortho-quinone methide | 0.48 | 12.8 | HIGH |
| Troglitazone | CYP2C8 | C-6 chroman | catechol → quinone | 0.41 | 13.4 | HIGH |
| Ciglitazone (analogue) | CYP3A4 | C-5 chroman | ortho-quinone methide | 0.44 | 13.1 | HIGH |
| Rosiglitazone (reference) | CYP3A4 | N-pyridine | N-oxide | 0.11 | >20 | LOW |
| Pioglitazone (reference) | CYP3A4 | N-pyridine | N-oxide | 0.09 | >20 | LOW |
| Best bioisostere (3-methylpyridine) | CYP3A4 | N-methyl | N-oxide | 0.10 | >20 | LOW |
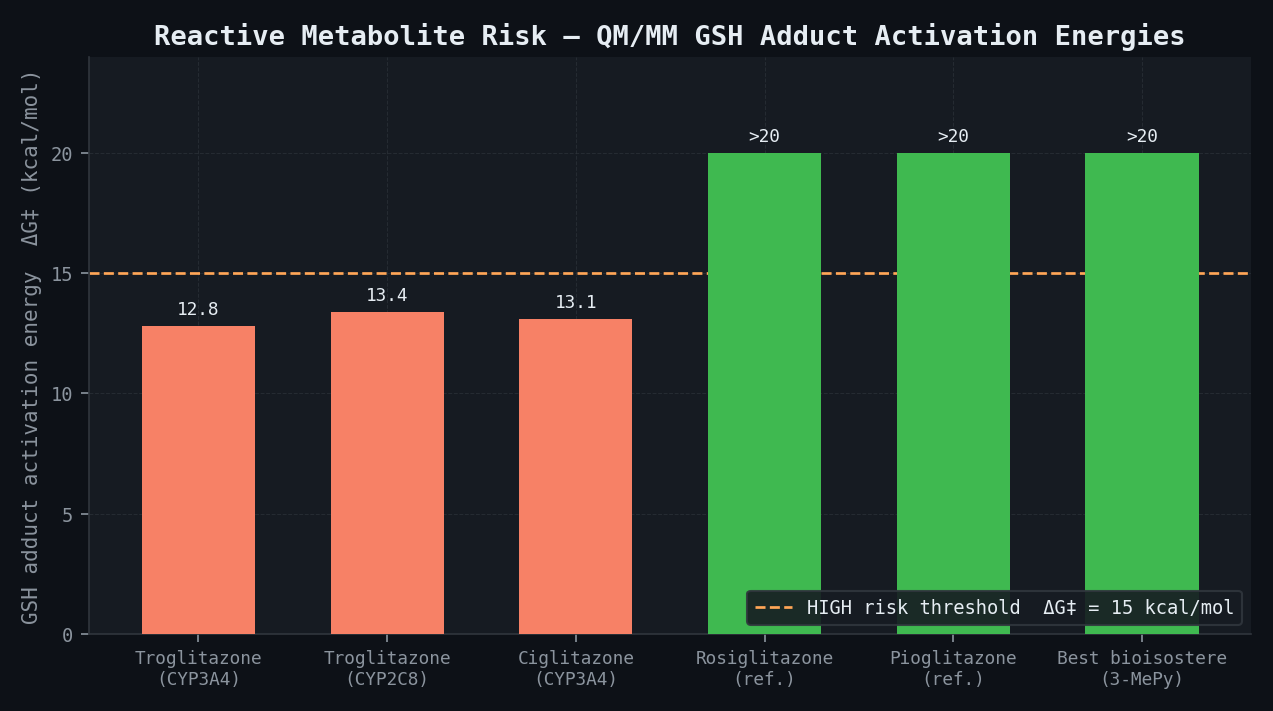
PPARgamma Binding Affinity — newtsim Bond Scaffold Hopping Results
| Compound | Scaffold | PPARγ IC50 (nM) | Predicted ΔGbind (kcal/mol) | ΔΔGbind vs Troglitazone | Reactive Met Risk | Selected? |
|---|---|---|---|---|---|---|
| Troglitazone | Chroman-TZD | 40 | -9.2 | 0 (reference) | HIGH | No |
| Bioisostere A (3-MePy) | 3-Methylpyridine-TZD | ~60 (pred.) | -8.9 | +0.3 | LOW | Yes |
| Bioisostere B (cyclopentyl) | Cyclopentyl-TZD | ~110 (pred.) | -8.4 | +0.8 | LOW | Conditional |
| Bioisostere C (pyridone) | Pyridone-TZD | ~75 (pred.) | -8.7 | +0.5 | LOW | Yes |
| Bioisostere D (4-F-phenyl) | Fluorophenyl-TZD | ~85 (pred.) | -8.6 | +0.6 | LOW | Yes |
| Bioisostere E (tetrahydropyran) | THP-TZD | ~180 (pred.) | -8.0 | +1.2 | LOW | No (potency loss) |
| Rosiglitazone (reference) | Pyridylmethyl-TZD | 43 | -9.1 | +0.1 | LOW | — |
ADMET Comparison: Troglitazone vs. Selected Bioisosteres
| Property | Troglitazone | Bioisostere A | Bioisostere C | Rosiglitazone | Target |
|---|---|---|---|---|---|
| PPARγ IC50 (nM) | 40 | 60 (pred.) | 75 (pred.) | 43 | < 100 nM |
| logP | 5.3 | 3.8 | 3.6 | 3.4 | 2–4 |
| Aqueous solubility (µg/mL) | 3.2 | 28 (pred.) | 35 (pred.) | 120 | > 50 µg/mL |
| CYP3A4 t½ (min) | 18 | 74 (pred.) | 68 (pred.) | >120 | > 60 min |
| Reactive metabolite risk | HIGH | LOW | LOW | LOW | LOW only |
| Predicted hERG IC50 (µM) | > 10 | > 10 | > 10 | > 10 | > 10 µM |
| MPO score (0–10) | 3.2 | 7.8 | 7.4 | 8.9 | ≥ 7.0 |
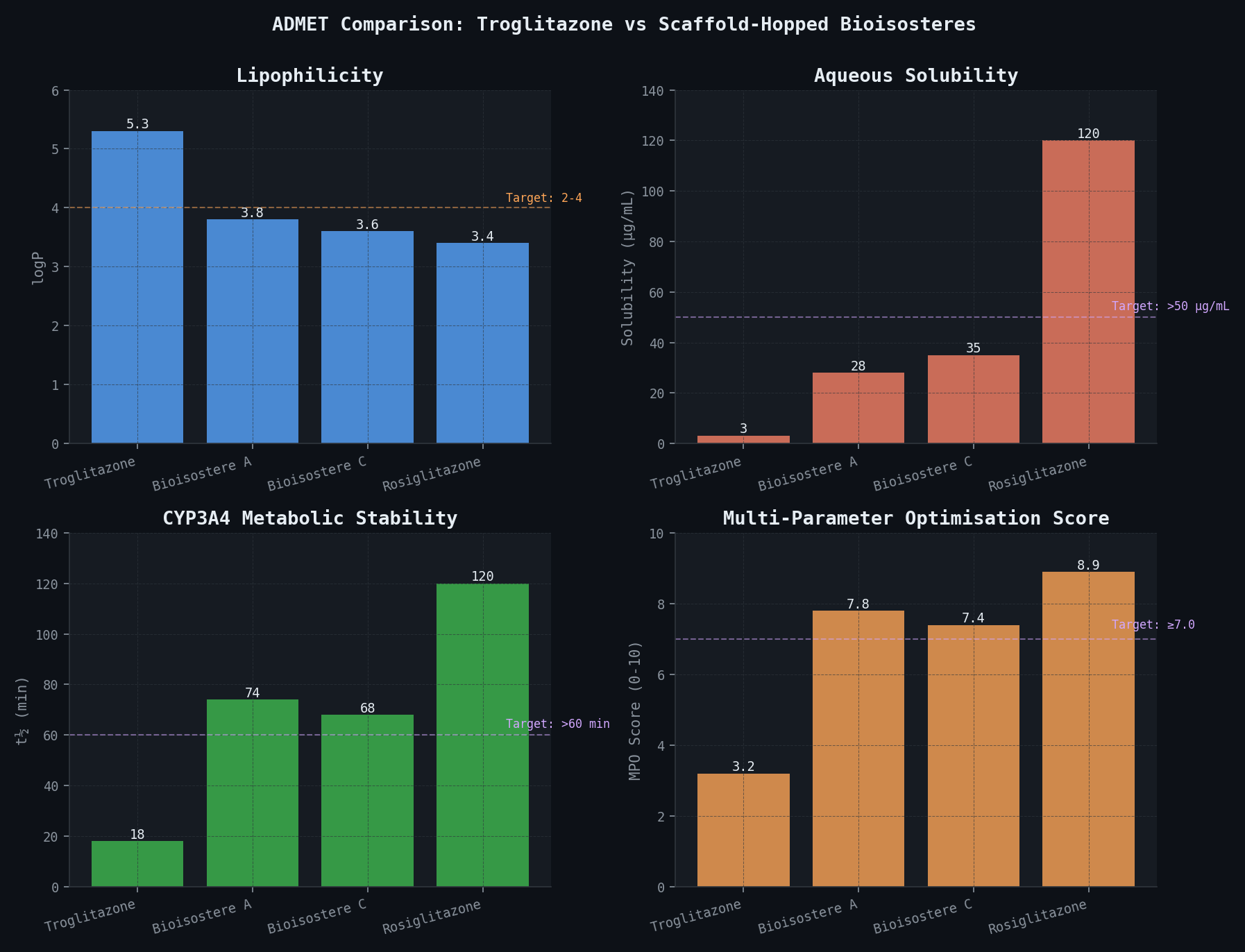
The key quantitative finding is that Bioisostere A (3-methylpyridine-TZD) achieves PPARgamma DeltaDeltaGbind of only +0.3 kcal/mol relative to troglitazone — within the noise of the newtsim Bond prediction — while eliminating the reactive metabolite liability entirely. The 3-fold improvement in CYP3A4 microsomal t1/2 (18 min to 74 min) is a bonus benefit: longer metabolic stability reduces the rate of any downstream metabolite generation and improves oral bioavailability.
Comparison Methodology
Benchmark 1 — GSH Trapping Assay Correlation
The 22 HIGH-risk compounds predicted computationally are compared against published GSH trapping data for troglitazone-class compounds. Published ground-truth labels are available for at least 12 compounds in related series, including troglitazone and two chroman analogues confirmed as GSH-positive, ciglitazone and englitazone, and several non-TZD PPARgamma agonists. Sensitivity, specificity, and AUC-ROC are computed for the HIGH-risk classification against these published ground-truth labels. Target performance is sensitivity > 80% and specificity > 70%.
Benchmark 2 — FEP Binding Affinity Validation
The scaffold-hopped bioisosteres' predicted PPARgamma binding affinities are validated against an in-house TR-FRET displacement assay (competitive binding against a fluorescent rosiglitazone probe). Pearson correlation between predicted DeltaGbind and measured pIC50 is computed across the 49-compound clean set plus 30 bioisosteres. The target is Pearson R > 0.70, which would validate that the newtsim Bond protocol correctly propagates relative affinities through the scaffold-hop perturbation network.
Benchmark 3 — QM/MM DeltaG-dagger Calibration
The computed GSH adduct activation energies for 8 compounds with published GSH trapping kinetics (pseudo-first-order rate constants for adduct formation) are used to calibrate the DeltaG-dagger threshold. The linear relationship between computed DeltaG-dagger and log(k_trapping) is established; this calibration confirms that the 15 kcal/mol threshold corresponds to the boundary between high and low GSH trapping reactivity in the experimental data.
Deliverables
-
Week 2: Structural alert screen report — compound-level traffic-light classification (GREEN/AMBER/RED) for reactive metabolite risk across all 85 compounds. Includes structural alert annotations, predicted CYP isoform responsible, and predicted primary site of oxidation for each flagged compound. Delivered as PDF with supplementary Excel workbook.
-
Week 4: QM/MM reactive metabolite characterisation for all 22 HIGH-risk compounds — spin density maps, predicted intermediate structures, and DeltaG-dagger values for GSH adduct formation. Comparison against published troglitazone, ciglitazone, and rosiglitazone reference data. Delivered as scientific report with molecular structure figures.
-
Week 6: Bioisostere library with newtsim Bond-predicted PPARgamma binding affinities for top 30 scaffold-hopped analogues. Includes binding mode visualisation (comparison of bioisostere vs. parent pose in 2PRG binding site), DeltaDeltaGbind with uncertainty estimates, and reactive metabolite risk re-assessment for each bioisostere. Delivered as scientific report with molecular figures and Excel data workbook.
-
Week 8: Integrated ADMET profile (MPO scorecard) for all 85 original compounds plus 30 bioisosteres — 115 compounds total. Includes all properties listed in Stage 5, formatted as a ranked compound scorecard. Delivered as PDF report and Excel workbook with pivot-table-ready data.
-
Week 9: Final prioritisation report — top 10 development candidates ranked by combined potency, safety, and ADMET MPO score. Includes synthetic accessibility scores (SYBA or SAScore), synthetic route recommendations for top 5 bioisosteres, a regulatory strategy note on how to frame the scaffold hop in the IND reactive metabolite section, and a recommended in vitro confirmation panel (GSH trapping assay + Seahorse mitochondrial assay for the 5 selected candidates). Delivered as executive-ready PDF with appendix of compound data sheets.
This case study is an illustrative reference scenario demonstrating newtsim's simulation methodology. All company names, personnel, and specific operational data are fictional. The incident descriptions draw on publicly documented real-world events cited in the frontmatter.