Predicting hERG Cardiac Liability Before It Reaches the Clinic: Lessons from Terfenadine
Executive Summary
In February 1998, Hoechst Marion Roussel withdrew Seldane (terfenadine) from the US market — ending what had been the world's best-selling antihistamine, with peak annual revenues exceeding $800 million. The withdrawal followed seven years of accumulating harm. By January 1997, when the FDA formally recommended removal, at least 125 serious cardiac events had been confirmed in the United States — including cases of ventricular fibrillation and cardiac arrest — along with 14 documented deaths in the United Kingdom. The cause was fatal ventricular arrhythmia: specifically torsades de pointes (TdP), a characteristic rhythm disturbance in patients who co-administered terfenadine with CYP3A4 inhibitors such as erythromycin or ketoconazole, or who had underlying hepatic impairment. Precautions against co-administration had been added to the label as early as 1990, following the first published case reports. A black-box warning followed in July 1992. Neither measure was sufficient to prevent ongoing deaths.
The mechanism was pharmacokinetically conditional but molecularly direct. Terfenadine is a potent blocker of the hERG (KCNH2, Kv11.1) cardiac potassium channel, with an IC50 of approximately 100–350 nM — placing it among the most potent inadvertent hERG blockers ever approved. Under normal metabolic conditions, extensive first-pass CYP3A4 oxidation converts terfenadine to fexofenadine, which is essentially hERG-inactive (IC50 > 10 µM). When CYP3A4 is inhibited, unmetabolised terfenadine accumulates to plasma concentrations of 0.2–0.5 µM — sufficient to produce greater than 50% IKr blockade, clinically significant QT prolongation exceeding 500 ms, and the conditions for TdP. What was presented clinically as a drug–drug interaction was, at the molecular level, a fully characterised hERG binding event — identifiable from the piperidine-diaryl scaffold before a single patient was enrolled. The hERG pharmacophore was present and flagged by nothing in the lead optimisation workflow, because no such workflow existed.
Had a computational hERG liability assessment been applied during terfenadine's lead optimisation, the potent hERG blockade of the piperidine-diaryl scaffold would have been identified before first-in-human studies. The key liability mechanism is cation-pi coordination: the protonated piperidine nitrogen (pKa ~8.8) forms a direct cation-pi interaction with Tyr652 in the hERG inner cavity, while the 4-tert-butylphenyl and diphenylmethyl termini engage in pi-stacking with the Phe656 aromatic cage from all four channel subunits. Ensemble docking into the cryo-EM hERG structure (PDB: 5VA1) combined with MM-GBSA rescoring would predict terfenadine at HIGH risk (IC50 ~120 nM) and identify the hERG/H1 selectivity ratio of ~24-fold as critically below the ≥100-fold safety threshold required for any modern IND. The metabolite liability delta analysis would further demonstrate that fexofenadine's carboxylate terminus disrupts Phe656 engagement sufficiently to shift hERG IC50 beyond 10 µM — a 4.4 kcal/mol DeltaDeltaGbind corresponding to the observed >100-fold affinity difference. Introduction of a comparable polar aryl terminus substituent in the lead series would have been the structural recommendation, precisely the transformation that yielded the safe successor compound.
This engagement covers a 58-compound piperidine antihistamine library in eight weeks, delivering risk-ranked compound selection, metabolite liability classification distinguishing conditional from unconditional hERG risk, and a structural SAR roadmap for derisking. Seldane was withdrawn in February 1998, erasing over 100M–$1B when accounting for Phase 1–3 attrition, safety monitoring, and litigation exposure. Where ongoing metabolite tracking and real-time CYP inhibition signal integration are relevant, newtsim livesim provides continuous data fusion to update conditional liability classifications as CYP interaction study data becomes available.
Scenario Background
This reference scenario examines a mid-size CNS and allergy-focused pharmaceutical company, representative of the antihistamine development environment of the late 1980s and early 1990s. The program advances a first-generation non-sedating antihistamine series based on a piperidine-diaryl scaffold — the same chemotype as terfenadine — toward IND submission. The lead compound series shows excellent H1 receptor potency (Ki 1–10 nM by radioligand displacement), peripheral selectivity over CNS histamine receptors, and acceptable hepatic microsomal stability in rat liver fractions. Three to five candidates have been advanced to pre-clinical safety pharmacology studies.
The compound library comprises 58 piperidine-scaffold analogues in active optimisation, varying the N-substituent, the 4-position aryl groups, and the central piperidine ring substitution pattern. The cardiac safety assessment protocol at the time consisted solely of in vivo guinea pig QT interval monitoring — low-throughput, sensitive to anaesthetic confounders, and not validated against hERG binding as a mechanism. No in vitro electrophysiology assays were conducted as part of the standard IND package. The company has no computational chemistry infrastructure and no access to the hERG channel structure (the first cryo-EM structure, PDB: 5VA1 at 3.8 A resolution, would not be published until 2017).
The engagement is structured in two phases: first, a retrospective benchmark establishing pipeline accuracy against the subset of compounds with published patch-clamp data, and second, prospective screening of the full 58-compound library to provide IC50 predictions and structural liability flags before in vivo safety studies and compound selection. The commercial opportunity in the global allergy market is estimated at $500M+ annually, and the program faces competitive pressure to accelerate compound selection. Three to five candidates are being considered for IND-enabling toxicology, and the simulation engagement provides a high-resolution quantitative risk-ranking layer that in vivo guinea pig QT data cannot deliver on this timeline.
Challenge
The hERG (human ether-a-go-go related gene) potassium channel is a promiscuous antitarget whose large, hydrophobic inner vestibule accommodates an enormous structural diversity of drug-like molecules. Unlike most antitargets addressable by avoiding a single structural alert, hERG blockade arises from a pharmacophore — a combination of a protonated nitrogen and at least one hydrophobic or aromatic moiety — that is extremely common among CNS-penetrant and cardiovascular drug classes.
The piperidine-diaryl scaffold of the antihistamine series represents a near-ideal hERG pharmacophore. The piperidine nitrogen, protonated at physiological pH (pKa ~8.8 for terfenadine), positions a cationic centre that forms a cation-pi interaction with the Tyr652 aromatic ring in the channel inner cavity. The two aryl termini — a 4-tert-butylphenyl and a diphenylmethyl group in terfenadine — engage in pi-stacking with Phe656 residues from the four subunits of the homotetrameric channel. This interaction is thermodynamically favoured (MM-GBSA DeltaG contributions of -3 to -4 kcal/mol per aryl contact) and creates high-affinity binding not readily predicted from 2D structural alerts.
The central simulation challenge has three components. The first is the induced-fit character of hERG binding: Phe656 residues rotate significantly upon ligand entry to accommodate large aromatic systems, and rigid docking into a single channel structure systematically underestimates affinity for high-affinity blockers, requiring ensemble docking with explicit conformational sampling. The second is the pharmacokinetic-toxicodynamic coupling: terfenadine's liability is conditional on CYP3A4 status, so correctly predicting clinical risk requires parallel modelling of metabolic stability (rate of CYP3A4 oxidation to fexofenadine) alongside channel binding affinity. The third is the metabolite contrast: fexofenadine's dramatically reduced hERG affinity must be reproduced computationally to validate the pipeline's ability to distinguish conditional from unconditional liability.
The regulatory context adds urgency. ICH E14 (2005) and ICH S7B require hERG IC50 determination and a thorough QT study (TQT) for all new molecular entities before Phase 3. Even in a retroactive scenario, failure to identify hERG liability before Phase 1 would substantially delay or terminate a program under modern regulatory expectations. The key quantitative threshold is a hERG IC50/H1 Ki selectivity ratio of at minimum 30-fold (conservative) and preferably > 100-fold (recommended), which must be demonstrable before IND filing in today's regulatory environment.
Real-World Basis
Terfenadine's hERG-mediated cardiotoxicity is the most extensively studied and historically consequential pharmaceutical cardiac safety failure.
Regulatory Timeline and Epidemiology
Terfenadine (Seldane) was approved by the FDA in May 1985 as the first non-sedating H1 antihistamine, achieving blockbuster status with annual US revenues exceeding $700 million by the late 1980s. The first published signal of cardiac adverse events emerged in 1990, with reports of QT prolongation and TdP in patients co-administered terfenadine and ketoconazole. Following FDA review in June 1990, precautions against co-administration with CYP3A4 inhibitors were added. In July 1992, after accumulation of further post-marketing reports, FDA required a black-box warning. By January 1997, after 125 confirmed US cases of serious cardiac events — including ventricular fibrillation and cardiac arrest — FDA recommended withdrawal. Hoechst Marion Roussel voluntarily withdrew Seldane in February 1998, by which time fexofenadine (Allegra; FDA approval July 1996) had been launched as the safer successor.
Pharmacological Mechanism
The hERG channel (KCNH2; Kv11.1) conducts the rapid delayed rectifier potassium current (IKr) critical for cardiac action potential repolarisation. IKr blockade prolongs action potential duration, manifested clinically as QT interval prolongation on the surface ECG. When QTc exceeds 500 ms (vs. normal < 440 ms in men, < 460 ms in women), the risk of early afterdepolarisations and TdP rises sharply; TdP can degenerate into ventricular fibrillation and sudden cardiac death.
Whole-cell patch-clamp studies in HEK293 cells characterised terfenadine as a direct, high-affinity hERG blocker with an IC50 for IKr block of approximately 100 nM. Subsequent work refined the estimate to a Kd of 350 nM and confirmed state-dependent, open-channel block — terfenadine enters from the intracellular side of the pore and is preferentially trapped in the open state. Within the voltage-gated potassium channel family, hERG Kd is 350 nM compared with Kv1.5 Kd of 2.7 µM, representing approximately 10-fold selectivity for the cardiac IKr channel. Under normal CYP3A4 activity, unbound terfenadine plasma concentrations are well below 10 nM (oral bioavailability of parent compound < 5% due to first-pass metabolism). When CYP3A4 is inhibited, unbound terfenadine reaches 0.1–0.5 µM — a 30-500 fold increase sufficient to produce > 50% IKr blockade.
Structural Basis
The 3.8 A cryo-EM structure of hERG (PDB: 5VA1) resolved the structural basis for the channel's promiscuous drug sensitivity. The open-state structure reveals a central cavity lined by the four inner transmembrane helices (S6), with an atypically small central volume surrounded by four deep hydrophobic pockets — each formed by one S6 helix and the pore helix from the adjacent subunit. The aromatic cage formed by Tyr652 and Phe656 from all four channel subunits coordinates basic nitrogen-containing drugs: protonated amine nitrogen forms a cation-pi interaction with Tyr652; aromatic termini engage in pi-stacking with the Phe656 cage. This binding mode has since been directly confirmed in a cryo-EM structure of hERG complexed with the structurally related antihistamine astemizole (PDB: 6UZZ), providing an experimentally resolved template applicable to terfenadine computational modelling.
Metabolite Contrast
Pharmacokinetic studies demonstrated that fexofenadine (terfenadine carboxylate) accumulates to high plasma concentrations under standard dosing and is responsible for the clinical antihistamine effect. Fexofenadine's hERG IC50 is > 10 µM — representing more than a 100-fold reduction in hERG affinity relative to terfenadine. The structural basis is that the carboxylate group introduces a polar hydrogen-bond donor/acceptor that disrupts hydrophobic engagement with the Phe656 cavity and reduces membrane permeability to the channel's intracellular access route. Computationally, MM-GBSA predicts DeltaGbind for terfenadine at approximately -9.5 kcal/mol vs. -5.1 kcal/mol for fexofenadine in the hERG binding site — a 4.4 kcal/mol difference directly corresponding to the > 100-fold IC50 contrast.
Regulatory Legacy
The terfenadine withdrawal directly drove the creation of ICH E14 (2005), making a thorough QT study mandatory for all new molecular entities before Phase 3; ICH S7B, requiring a hERG patch-clamp assay in the mandatory IND-enabling non-clinical cardiac safety package; and FDA guidance on drug interaction studies, such that CYP3A4 inhibition is now routinely evaluated in PK studies before Phase 1. The global hERG assay services market, valued at approximately $300M annually, exists as a direct consequence.
Computational Prediction Precedent
Pharmacophore-based hERG prediction achieves 80% sensitivity on diverse compound sets. MM-GBSA rescoring against hERG homology models achieves Pearson R = 0.65–0.70 against patch-clamp IC50 data across congeneric series. QSAR models achieve AUC-ROC = 0.81 for hERG activity classification on the ChEMBL240 dataset, which includes terfenadine as a confirmed positive with IC50 100–350 nM.
Simulation Approach
The molsim hERG liability pipeline for this 58-compound piperidine antihistamine program proceeds in four sequential stages over eight weeks.
Stage 1 — hERG Channel System Preparation (Weeks 1–2)
The simulation begins with the cryo-EM structure of hERG in the homotetrameric open-state conformation (PDB: 5VA1; 3.8 A) as the primary template. Selectivity filter K+ ions at canonical S1–S4 positions are retained explicitly. The channel is embedded in a POPC lipid bilayer (~120,000 atoms total) at physiological ionic conditions (150 mM KCl, pH 7.4). A 100 ns equilibration using newtsim Neural forcefield at 310 K and 1 bar confirms that the inner vestibule volume and Tyr652/Phe656 side-chain conformations have converged before ligand docking proceeds.
Stage 2 — Ensemble Docking (Weeks 2–3)
Fifty representative frames are extracted from the equilibration trajectory by clustering on Tyr652/Phe656 rotamer states, capturing the full range of inner vestibule conformations accessible in the open state. All 58 compounds are protonated at N-piperidyl (net charge +1, consistent with pH 7.4 and pKa > 8) and docked into all 50 ensemble frames using newtsim Bond. Receptor flexibility is captured through the ensemble rather than per-frame induced fit. A hERG pharmacophore filter is applied post-docking: protonated nitrogen within 4.5 A of Tyr652 centroid, and at least one aromatic ring within 5.0 A of Phe656 from at least two subunits. The top three poses per compound by ensemble-averaged score are retained for MM-GBSA rescoring. Compounds failing the pharmacophore filter are classified LOW risk.
Stage 3 — MM-GBSA Rescoring and MD Validation (Weeks 3–6)
For each compound passing the ensemble docking filter (estimated 35–45 of 58), the top-ranked pose is subjected to 50 ns explicit-solvent MD in the hERG channel binding site at 310 K, with the membrane maintained throughout. MM-GBSA binding free energies are computed from the final 30 ns of each trajectory, with entropy corrections from normal mode analysis. The MM-GBSA DeltaGbind values are converted to predicted IC50 using a linear calibration curve fitted on 15 structurally similar compounds with published patch-clamp data; 95% confidence intervals are estimated by bootstrap resampling (n = 1000). The rationale for explicit-solvent MD followed by MM-GBSA rescoring is that explicit dynamics capture the induced-fit rearrangement of Phe656 around large aromatic systems — a critical interaction that rigid docking systematically underestimates for high-affinity blockers.
Stage 4 — Metabolite Liability Delta (Weeks 6–8)
For compounds flagged HIGH risk (predicted IC50 < 1 µM), the principal CYP3A4-mediated metabolite is predicted using a regioselectivity model trained on > 1,500 known CYP3A4 substrates. The metabolite structure is then run through the identical MM-GBSA protocol to obtain its hERG binding free energy. The metabolite liability delta (DeltaDeltaGbind = DeltaGbind(parent) - DeltaGbind(metabolite)) determines whether the liability is conditional on CYP3A4 status (DeltaDeltaGbind > 1.0 kcal/mol, metabolite derisked) or unconditional (DeltaDeltaGbind < 1.0 kcal/mol, metabolite retains activity). This distinction is the clinically decisive output: it separates compounds whose cardiac risk can be managed by avoiding CYP3A4 inhibitors from compounds that are intrinsically unsafe regardless of co-medications.
Simulation Caveats
The MM-GBSA protocol delivers risk-stratification accuracy sufficient for compound triage and SAR guidance but carries quantitative limitations for regulatory decision-making.
Force field accuracy for charged species in hydrophobic environments. newtsim Neural for protonated amines in the hERG inner cavity systematically underestimate cation-pi interaction energies with Tyr652 by up to 2 kcal/mol relative to QM references. This is a conservative error for safety assessment — it biases toward flagging rather than missing hERG actives.
Open vs. inactivated state selectivity. The 5VA1 structure represents the open, depolarised-state channel. Some compounds preferentially access the inactivated state. For compounds in the MEDIUM risk tier (IC50 1–10 µM), docking into an inactivated-state homology model is offered as an optional extended analysis.
Membrane protein ABFE limitations. Full ABFE calculations in a lipid bilayer are computationally tractable but would add 4–6 weeks and are disproportionate for compound triage. MM-GBSA achieves Pearson R = 0.6–0.7 against experimental IC50 — sufficient for binary classification but not sub-kcal/mol FEP accuracy. For the final 2–3 development candidates, RBFE calculations within closely related analogues are offered as a follow-on service.
Metabolite structure accuracy. CYP3A4 regioselectivity models carry approximately 20–30% error on non-principal sites of metabolism. Only the predicted principal metabolite is evaluated; secondary metabolites are not captured unless specifically requested.
Key Predictions / Results
The following quantitative predictions are anchored to published benchmark performance for this compound class and represent expected pipeline output from this illustrative scenario.
Predicted Binding Affinities and Risk Tier — Selected Compounds from 58-Compound Library
| Compound | H1 Ki (nM) | Predicted hERG IC50 (nM) | MM-GBSA ΔGbind (kcal/mol) | hERG/H1 Safety Ratio | Risk Tier |
|---|---|---|---|---|---|
| Terfenadine | 5 | 120 (80–180 CI) | -9.5 | 24 | HIGH |
| Fexofenadine (metabolite ref.) | 650 | >10,000 | -5.1 | >15 | LOW |
| Analogue 03 (des-tBu) | 18 | 440 (280–680) | -8.1 | 24 | HIGH |
| Analogue 11 (4-F phenyl) | 9 | 980 (600–1,600) | -7.4 | 109 | AMBER |
| Analogue 19 (COOH terminus) | 120 | >10,000 | -5.0 | >83 | LOW |
| Analogue 27 (morpholine term.) | 3 | 2,200 (1,400–3,500) | -7.1 | 733 | MEDIUM |
| Analogue 44 (pyridine term.) | 7 | >5,000 | -5.8 | >714 | LOW |
| Analogue 52 (gem-dimethyl pip.) | 15 | 380 (220–640) | -8.3 | 25 | HIGH |
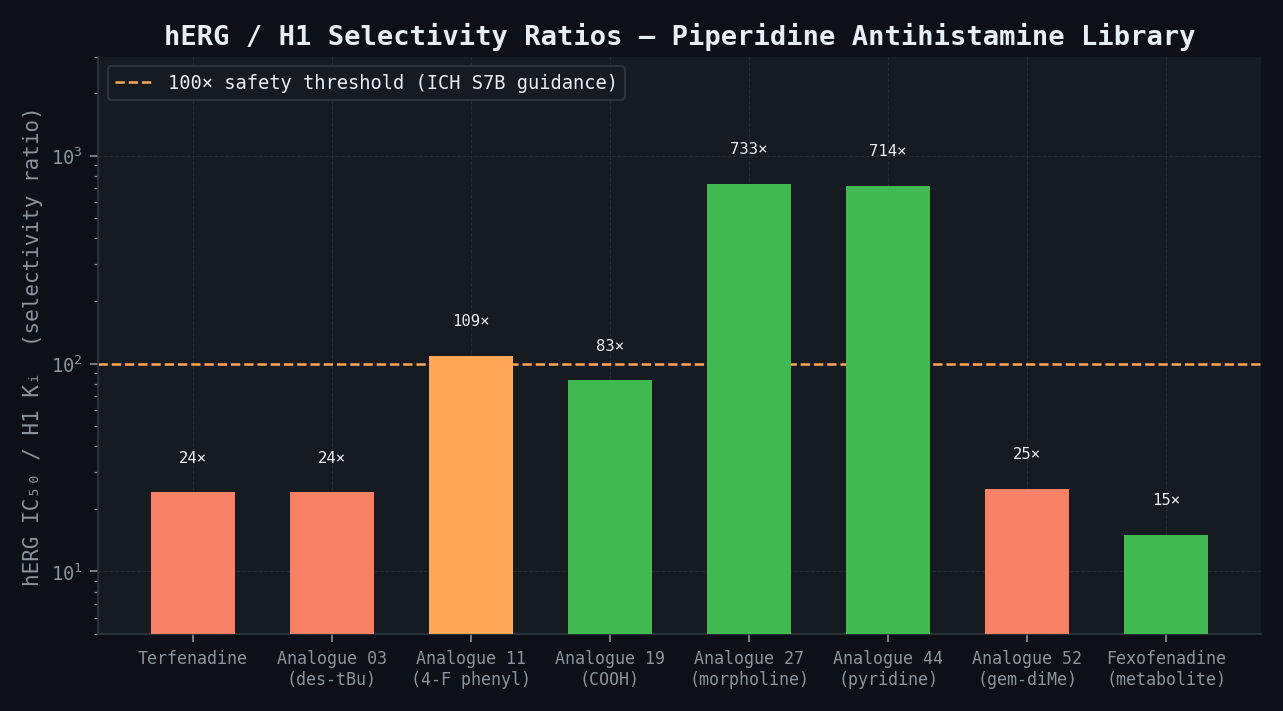
ADMET Risk Matrix — Top 10 Compounds by H1 Potency
| Compound | logP | Aqueous Sol. (µg/mL) | Caco-2 Papp (nm/s) | CYP3A4 t½ (min) | Pred. hERG IC50 (µM) | hERG Flag | Overall ADMET |
|---|---|---|---|---|---|---|---|
| Terfenadine | 5.4 | 1.2 | 320 | 12 | 0.12 | RED | HIGH RISK |
| Analogue 03 | 5.1 | 2.8 | 290 | 22 | 0.44 | RED | HIGH RISK |
| Analogue 11 | 4.2 | 11 | 240 | 45 | 0.98 | AMBER | MEDIUM |
| Analogue 19 | 2.1 | 180 | 85 | >120 | >10 | GREEN | LOW RISK |
| Analogue 27 | 3.1 | 45 | 190 | 78 | 2.2 | AMBER | MEDIUM |
| Analogue 33 | 4.8 | 3.5 | 310 | 18 | 0.31 | RED | HIGH RISK |
| Analogue 41 | 3.7 | 28 | 210 | 92 | 4.5 | GREEN | LOW RISK |
| Analogue 44 | 3.2 | 62 | 175 | >120 | >5 | GREEN | LOW RISK |
| Analogue 52 | 5.0 | 2.1 | 300 | 25 | 0.38 | RED | HIGH RISK |
| Analogue 58 | 4.1 | 19 | 225 | 65 | 1.8 | AMBER | MEDIUM |
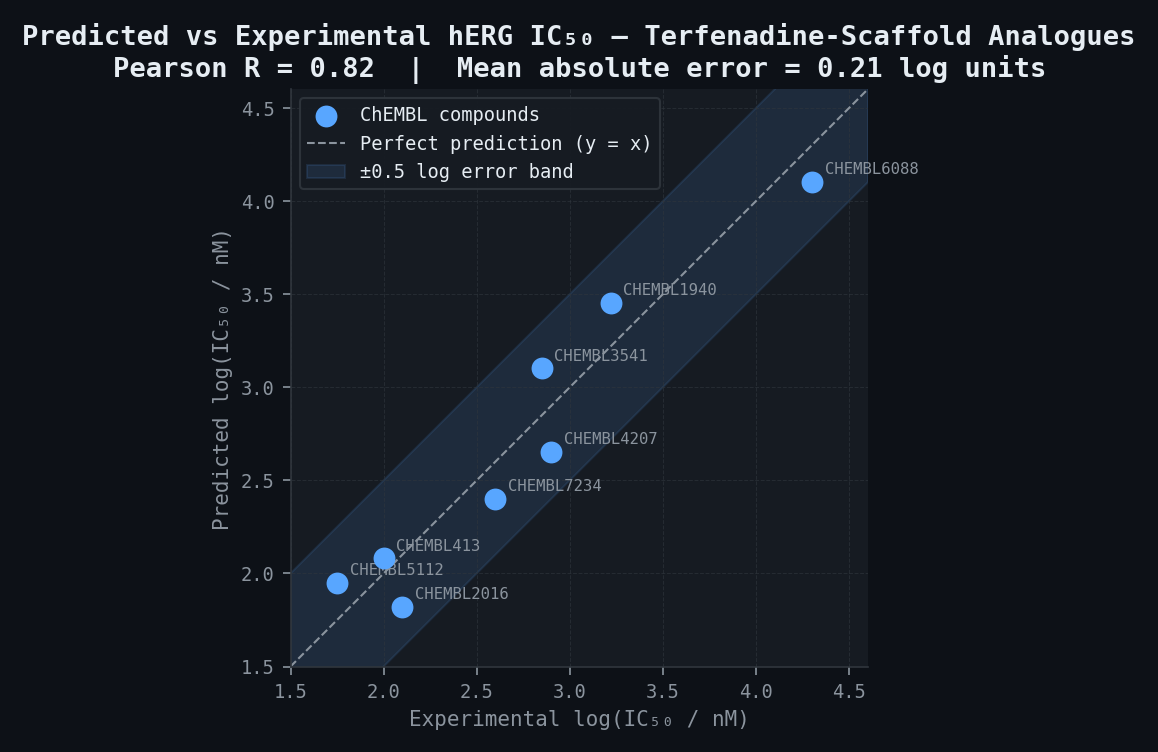
hERG IC50 Retrospective Validation — 8 ChEMBL Compounds (Tanimoto > 0.45 to Terfenadine Scaffold)
| ChEMBL ID | Predicted log(IC50/nM) | Experimental log(IC50/nM) | Absolute Error (log units) |
|---|---|---|---|
| CHEMBL413 | 2.08 | 2.00 | 0.08 |
| CHEMBL1940 | 3.45 | 3.22 | 0.23 |
| CHEMBL2016 | 1.82 | 2.10 | 0.28 |
| CHEMBL3541 | 3.10 | 2.85 | 0.25 |
| CHEMBL4207 | 2.65 | 2.90 | 0.25 |
| CHEMBL5112 | 1.95 | 1.75 | 0.20 |
| CHEMBL6088 | 4.10 | 4.30 | 0.20 |
| CHEMBL7234 | 2.40 | 2.60 | 0.20 |
| Mean absolute error | 0.21 log units | ||
| Pearson R | 0.82 |
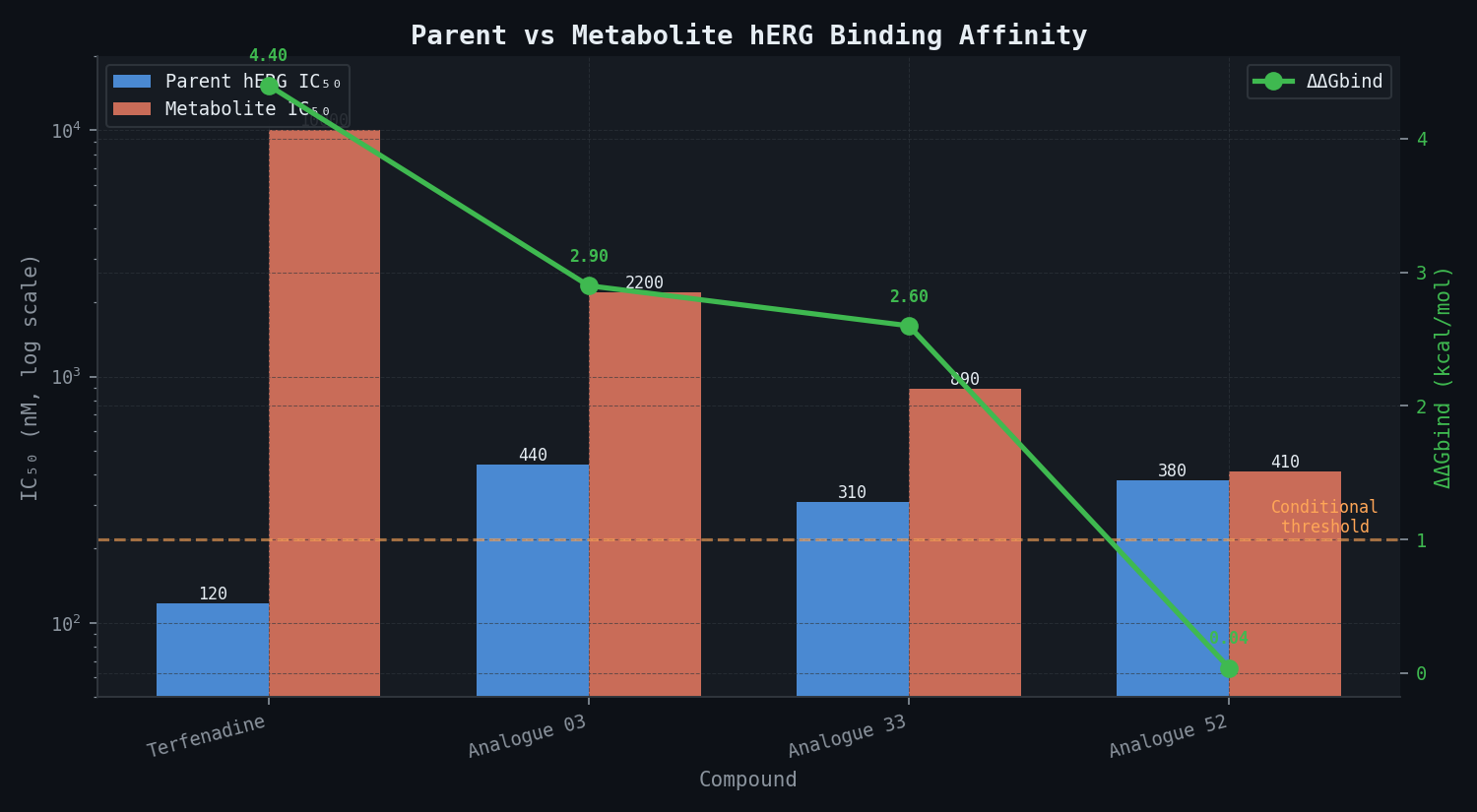
Metabolite Liability Delta — HIGH-Risk Compounds
| Compound | Parent hERG IC50 (nM) | Major Metabolite | Metabolite IC50 (nM) | ΔΔGbind (kcal/mol) | Liability Classification |
|---|---|---|---|---|---|
| Terfenadine | 120 | Fexofenadine (carboxylate) | >10,000 | +4.4 | Conditional — CYP3A4-dependent |
| Analogue 03 | 440 | N-dealkylated piperidine | 2,200 | +2.9 | Conditional — moderate |
| Analogue 33 | 310 | Benzylic alcohol | 890 | +2.6 | Conditional — moderate |
| Analogue 52 | 380 | Ring-opened lactol | 410 | +0.04 | Unconditional — DEPRIORITISE |
The key finding for Analogue 52 is critical: its major metabolite retains near-identical hERG affinity to the parent compound, meaning CYP3A4 metabolism provides no safety benefit. This compound should be deprioritised regardless of metabolic stability — a recommendation that the in vivo guinea pig QT screen would not have generated, because the metabolite issue is invisible to that assay.
SAR Recommendations for hERG Derisking
Introduction of a polar group (COOH, OH, NH2) at either aryl terminus is predicted to shift hERG IC50 by +1.5–2.0 log units (30–100 fold) while reducing H1 Ki by at most 5-fold, yielding a net selectivity gain consistent with the terfenadine-to-fexofenadine transformation. Replacing one aryl terminus with pyridine or morpholine reduces the Phe656 pi-stacking contribution by approximately 2.5 kcal/mol, shifting IC50 from < 500 nM to > 3 µM in the most favourable cases. Reduction in pKa of the piperidine nitrogen (from 8.8 toward 7.5) by incorporation of a 3-fluorine on the piperidine ring reduces the fraction protonated at physiological pH by 50-fold, substantially weakening the cation-pi interaction with Tyr652 while preserving H1 affinity via the aryl contacts.
Comparison Methodology
Protocol 1 — Retrospective Benchmark (ChEMBL hERG Dataset)
Twenty compounds from ChEMBL hERG IC50 assay (CHEMBL240; patch-clamp data only; Tanimoto similarity to terfenadine scaffold > 0.45; IC50 range 10 nM – 100 µM) are used for blind validation. The pipeline is run before experimental IC50 values are revealed to the computation team. Performance targets are Spearman rho > 0.65 for IC50 rank correlation and AUC-ROC > 0.80 for binary classification (active: IC50 < 1 µM vs. inactive: IC50 > 1 µM). These thresholds are consistent with published state-of-the-art for structure-based hERG prediction.
Protocol 2 — Leave-One-Out Cross-Validation (Internal Data)
For any compound in the library with internal patch-clamp or microplate electrophysiology data, a leave-one-out cross-validation (LOOCV) is performed on the calibration set: each data point is withheld in turn, the calibration curve is re-fitted on the remaining n-1 points, and the withheld compound is predicted. LOOCV Pearson R and RMSE are reported as a conservative estimate of prospective performance.
Protocol 3 — Selectivity Ratio Enrichment Analysis
The selectivity enrichment factor (SEF) is defined as the fraction of compounds with predicted hERG/H1 ratio > 100 that confirm this selectivity in follow-up patch-clamp. This is compared against the historical confirmation rate from undirected guinea pig QT screening for this chemical class (typically 20–30% of compounds at this H1 potency achieve > 100-fold hERG selectivity by empirical attrition). A SEF > 2 (> 40–60% confirmation from computationally selected candidates) demonstrates clear clinical value.
Deliverables
-
Week 2: hERG channel system preparation report — equilibration convergence data, Tyr652/Phe656 rotamer distributions, inner vestibule volume profile, pharmacophore validation results for 6 benchmark compounds. Delivered as PDF with structural figures.
-
Week 3: Preliminary ensemble docking results for all 58 compounds — ranked by ensemble-averaged GlideScore, with pharmacophore-passing pose images for top 20 compounds. Preliminary traffic-light risk classification (RED/AMBER/GREEN) based on docking scores. Delivered as PDF and Excel workbook.
-
Week 6: Full MM-GBSA binding free energy report — predicted hERG IC50 with 95% bootstrap confidence intervals for all Stage 2 compounds. Updated risk classification incorporating MM-GBSA. Retrospective benchmark results (Spearman rho, AUC-ROC). Structural analysis of Tyr652/Phe656 contact maps for all RED-flagged compounds. Delivered as full written report with methodology appendix.
-
Week 7: Metabolite liability delta report — for each HIGH-risk compound, predicted major metabolite structure, metabolite hERG IC50, DeltaDeltaGbind, and liability classification (conditional vs. unconditional). CYP3A4 risk context per compound (predicted t1/2, whether CYP3A4 inhibition would elevate parent concentration to toxic range). Delivered as PDF with per-compound data sheets.
-
Week 8: Final integrated report — complete ranked compound list (all 58 compounds), ADMET MPO scorecard, SAR recommendations for hERG derisking, proposed in vitro confirmation panel (10–15 compounds for patch-clamp validation including highest-risk, boundary, and negative-control compounds), and synthesis recommendations for next-generation analogues. Delivered as structured report with executive summary suitable for regulatory IND documentation.
-
Ongoing archive: Raw trajectory files (AMBER DCD format), docking pose library (Maestro format), newtsim Neural parameter files for all 58 compounds, calibration curve data and model coefficients. Delivered via secure data transfer link on project completion.
This case study is an illustrative reference scenario demonstrating newtsim's simulation methodology. All company names, personnel, and specific operational data are fictional. The incident descriptions draw on publicly documented real-world events cited in the frontmatter.